



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
完全性雄激素不敏感综合征合并膀胱瘘1例报道及文献回顾
[花克涵1 , 杨磊2 , 张晓威1 , 白文俊1 , 李清1 , 徐涛1, △  ]
]
]
|
|


雄激素不敏感综合征(androgen insensitivity syndrome,AIS)是一种罕见遗传病。由于雄激素受体(androgen receptor,AR)基因的突变导致其对雄激素产生抵抗及不应答,男性生殖系统出现不同程度异常发育并呈现出女性表型。手术切除隐睾及不同类型成形术可纠正畸形,激素替代治疗也是术后康复的关键。本文报告1例43岁患者,社会性别为女性,初因反复腹水及间断腹痛原因待查收入消化内科。体格检查、超声、核型分析、激素水平等检验检查符合完全性AIS。转入泌尿外科行隐睾切除术,术中发现膀胱顶壁紧邻脐中韧带与膀胱交界处有一瘘道,遂明确腹水原因,予切除并送病理。病理结果提示睾丸组织Sertoli细胞腺瘤及Leydig细胞巢状增生,膀胱瘘口周围组织非典型增生。出院后予雌激素替代治疗。睾丸切除术后雄激素合成障碍导致促性腺激素、雌孕激素水平发生变化,后续雌激素替代治疗联合心理辅导可稳定激素水平并提高生活质量。患者有可疑家族史,系谱图分析为X连锁隐性遗传,但尚不能明确。对于膀胱顶壁瘘的成因本文提出3种假说:直接手术损伤、癌症局部复发以及脐尿管异常发育,但脐尿管发育异常与本病是否存在相关性仍需进一步研究。
Androgen insensitivity syndrome (AIS) is a very uncommon genetic disorder that results from the resistance of androgen receptor (AR) to androgen, which influences the formation of the male genitalia and in turn presents with female phenotype. Surgical resection of undesceaded testicle and different kinds of genitoplasty are crucial methods to correct the deformity of reproductive system, as well as hormone replacement therapy, which is an essential therapy for postoperational rehabilitation in AIS patients. A 43-year-old patient, who was socially female, was first admitted to gastroenterology department due to recurrent ascites and occasional abdominal pain with unknown origin. Taking physical examination, ultrasonography, karyotype analysis and sex hormone levels into consideration, the overall manifestations revealed the typical clinical features of complete androgen insensitivity syndrome. After that she was transferred to urology department for laparoscopic gonadectomy. During the surgery, doctors found that there was a vesical fistula on the upper wall near the conjunction between the bladder and ligamenta umbilicale medium, which explained the recurrent ascites for more than 4 years. After resecting the testicles and the tissues around the vesical fistula for histopathology, the result suggested Sertoli cell adenoma, hyperplastic Leydig cells and urothelium atypical hyperplasia. Hormone replacement therapy was given right after discharge. The hormone levels of follicle-stimulating hormone, luteinizing hormone, estradiol and progesterone were modulated by the dysfunction of androgen production after gonadectomy and hormone replacement therapy together with psychotherapy could stabilize her hormone levels and improve the quality of her life. The patient was suspicious of AIS family history and the pedigree was made to analyze her family which was possibly X-linked recessive pattern. We propose three possible hypotheses of the fistula, which are direct surgical injury, recurrence of bladder cancer and congenital urachal anomalies. But whether it is relevant between urachal anomalies and AIS is yet to be discovered.
雄激素不敏感综合征(androgen insensitivity syndrome, AIS), 又称为睾丸女性化, 是一种罕见的X连锁隐性遗传病。由于X染色体上编码雄激素受体(androgen receptor, AR)的基因发生突变, AR产生功能异常, 产生一系列雄激素抵抗综合征, 靶器官对雄激素不敏感或部分敏感, 导致遗传性别为男性的患者出现不同程度的女性化, 主要表现在第二性征及内外生殖器, 形成性别与性征的不匹配[1, 2]。根据AR对雄激素不同程度的敏感度, AIS可以分为完全性AIS(complete AIS, CAIS)、部分性雄激素不敏感综合征(partial AIS, PAIS)及轻型AIS(mild AIS, MAIS)[3]。AIS在遗传性别为男性的人群中发病率为1/100 000~1/90 000, CAIS的发病率在男性新生儿中为1/64 000~1/20 000[4]。目前较为主流的治疗方法为睾丸切除术[5], 并在术后予激素替代治疗, 两者缺一不可, 以建立良好的性别认知并促进身心健康的恢复[2]。本文将报道1例CAIS合并膀胱瘘的病例。
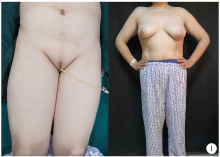
患者43岁, 社会性别女, 未婚。因“ 间断腹痛、腹胀4年余” 收住北京大学人民医院消化内科, 既往8年前曾行经尿道膀胱肿瘤电切术(transurethral resection of the bladder tumors, TURBt)并行术后膀胱药物灌注化疗2年, 发现高血压、脂肪肝、多发肾囊肿2年。患者原发性闭经, 未婚未育, 其二姐及两位姨妈均原发性闭经、未育, 且无阴毛、腋毛发育。患者体重80 kg, 身高170 cm, 体重指数 27.68 kg/m2, 属于肥胖。患者女性面容, 无明显胡须及喉结, 无腋毛发育; 外阴外观女性, 有阴道及尿道发育, 无阴毛发育, 双侧乳房发育正常(图1); 双侧腹股沟区未触及包块。
 | 图1 乳房及阴毛发育Figure 1 Breast and pubic hair development |
辅助检查:(1)性激素测定提示性腺功能不全:卵泡刺激素(follicle-stimulating hormone, FSH)68.14 IU/L(正常值:卵泡期3.85~8.78 IU/L, 绝经期16.74~113.59 IU/L; 男性1.27~19.26 IU/L); 促黄体生成素(luteinizing hormone, LH)31.67 IU/L(正常值:卵泡期2.12~10.89 IU/L, 绝经期10.87~58.64 IU/L; 男性1.24~8.62 IU/L); 雌二醇29.08 ng/L(正常值:卵泡期27~122 ng/L, 绝经期≤ 40 ng/L; 男性≤ 47 ng/L); 孕激素0.73 μ g/L(正常值:卵泡期0.31~1.52 μ g/L, 绝经期≤ 0.78 μ g/L; 男性0.14~2.06 μ g/L); 睾酮4.13 nmol/L(女性≤ 2.60 nmol/L; 男性6.07~27.10 nmol/L)。(2)染色体核型(G显带): 46, XY。(3)超声:双侧腹股沟条状低回声, 与腹腔相通, 达右侧大阴唇旁, 其内椭圆形无回声, 范围3.0 cm× 1.1 cm, 无回声旁见圆形低至无回声结构, 大小1.0 cm× 0.9 cm, 未见明显彩色血流信号且超声造影未见明显造影剂灌注, 考虑类似睾丸结构。(3)增强CT:子宫显示不清(发育不全?), 双侧腹股沟管内可见软组织密度影, 盆腔可见少量水样密度影。考虑假两性畸形合并CAIS可能性大。
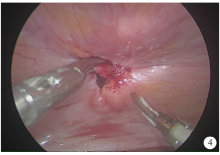
患者为求进一步诊疗转诊至我院泌尿外科。由于高度怀疑腹股沟内为睾丸组织(隐睾), 且因患者年龄较大, 睾丸组织易癌变, 遂行腹腔镜下双侧隐睾切除术。术者取脐上弧形切口以及左、右下腹各一横切口置入穿刺器, 在气腹条件下探查腹腔, 于右侧内环口处见萎缩睾丸组织, 左侧睾丸组织不明显(图2、3), 分离并取出双侧睾丸组织, 关闭内环口以避免腹股沟斜疝形成。随后探查腹腔内情况时发现膀胱顶壁一直径约0.3 cm的缺损(图4), 紧邻脐中韧带与膀胱连接处(图中术野未显露), 经尿道向膀胱向灌注生理盐水, 可见液体通过该瘘口流入腹腔, 证实瘘管存在, 符合既往反复腹水导致腹胀的病史。切除瘘口周围1 cm组织并封闭缝合, 取睾丸组织及瘘口旁切除的膀胱组织送病理检查。病理结果提示, 左、右侧睾丸可见结节状增生的肿物, 腺管增生, 内衬卵圆形细胞, 未见精母细胞及精子, 免疫组织化学染色结果为细胞角蛋白(cytokeratin, CK)(-)、波形蛋白(vimentin)(+)、上皮细胞膜抗原(epithelial membrane antigen, EMA)(-)、抑制素α (α -inhibin)(+)、钙视网膜蛋白(calretinin, CR)(+)、分化抗原簇99(cluster of differentiation 99, CD99)(+)、威尔姆瘤核蛋白1(Wilms’ tumor nuclear protein 1, WT-1)(+)、T细胞识别黑色素瘤抗原-1(Melan-A/MART-1, MelanAm)(-)、Ki-67(1%+), 符合Sertoli细胞腺瘤, 大小1.7 cm× 1.5 cm, 间质可见Leydig细胞巢状增生; 左、右侧可见囊肿, 被覆立方上皮, 大小分别为3.5 cm× 1.5 cm、2.0 cm× 1.8 cm; 膀胱顶壁可见灶状尿路上皮非典型增生(图5)。
 | 图2 切除的双侧隐睾Figure 2 Bilateral dissected testicles |
 | 图3 术中所见萎缩的睾丸组织Figure 3 Undescended and atrophic testicle tissue |
 | 图4 膀胱顶壁瘘Figure 4 The vesical fistula on the upper wall of bladder |
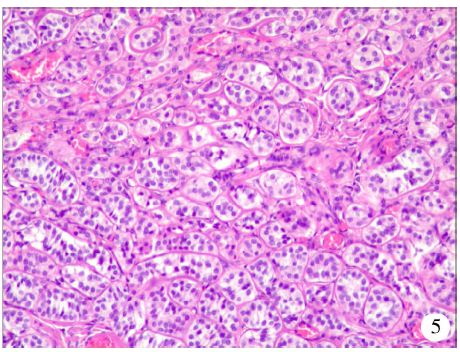 | 图5 睾丸组织HE染色(× 100)Figure 5 HE staining of testicular tissue (× 100) |
术后予对症支持治疗, 第12天出院, 嘱行激素替代治疗, 并定期于妇科及内分泌科门诊复查性腺激素水平以逐步调整激素剂量及给药方案。
AIS是一种少见的先天性性发育异常疾病, 主要因雄激素受体对雄激素的应答能力下降或丧失, 从而导致遗传性别为男性的个体在性征及生殖系统发育方面出现不同程度的女性化[1]。从分子机制上讲, AR是一种核受体, 属于配体依赖的转录因子超家族[6]。AR基因由8个外显子组成, 包含4个主要部分:由第1外显子编码的N-末端结构域(N-terminal domain, NTD), 由第2、3外显子编码的DNA结合结构域(DNA-binding domain, DBD), 铰链区和由第4~8外显子编码的C末端配体结合结构域(C-terminal ligand-binding domain, LBD), 4部分正常编码后形成AR[1, 7, 8]。睾酮或双氢睾酮(dihydrotestosterone, DHT)与AR结合后, 受体结构产生变化并移位进入细胞核中与DNA结合形成二聚体, 从而使靶细胞的相应基因编码、转录并翻译出相应的产物[9]。但若出现单一碱基对替换或无义点突变等将会造成AR基因功能缺陷, 影响转录及翻译过程, 最终将引起AR对雄激素的敏感度下降甚至丧失[10]。迄今为止, 研究人员已发现多达400个不同AR基因突变位点[11], 数据统计表明, 约90%~95%的CAIS是由于AR基因突变, 但仅有28%~50%的PAIS出现AR基因突变, 说明除AR基因突变外还有许多未知的分子生物学机制尚未被发现[12]。
从胚胎发育上讲, 性别分化发生于胚胎发育早期。正常男性胎儿在胚胎发育第6周, 由Y染色体性别决定基因(sex-determining region on the Y chromosome, SRY)编码的睾丸决定因子(testis determining factor, TDF)促进生殖嵴向睾丸发育; 第8周睾丸间质细胞(Leydig cells)开始分泌睾酮, 促进中肾管发育, 后者在第9周分化为内生殖器— — 附睾、输精管和精囊。值得一提的是, 睾酮最活跃的代谢产物DHT能促进生殖结节向阴茎转化以及阴茎腹侧缝的闭合, 同时对尿道海绵体部以及腹正中嵴的形成有着决定性作用。另外, 促性腺激素能够刺激睾丸支持细胞产生抗苗勒氏管激素(anti-Miillerian hormone, AMH), 抑制中肾旁管即苗勒氏管进一步发育为子宫、输卵管及阴道上1/3[1, 13], 上述激素共同作用完成正常胚胎发育过程。但由于AIS患者体内睾酮及DHT未能产生正常生理作用, 男性内外生殖器随即出现不同程度的发育障碍, 同时患者体内AMH分泌正常, 导致女性内外生殖器均不发育, 最终表现为CAIS、PAIS及MAIS的不同临床特点。
CAIS的典型临床表现是原发性闭经或婴幼儿腹股沟疝。除此之外, 因腹股沟区肿物或大阴唇肿胀而就诊者最终被确诊为隐睾, 亦或是发现遗传性别与生长发育表型不匹配而就医者也不在少数。CAIS患者的社会性别普遍为女性, 患者一般呈女性体态, 因体内高雄激素水平, 身材较一般女性高大但未达到男性水平; 乳房发育基本正常, 无腋毛及阴毛; 有阴道口, 但阴道为盲端且长度约为正常女性的2/3, 无子宫、卵巢及输卵管发育; 阴蒂较小, 大阴唇及小阴唇发育基本正常[2, 14, 15]。PAIS的临床表现通常趋向于多样化, 表现为男性内、外生殖器不同程度的女性化, 包括小阴茎、尿道下裂、阴囊对裂等[1]。MAIS患者通常表现为男性表型或仅有小阴茎畸形, 但很有可能在青春期出现乳房发育或在成年后出现不育症[11]。
诊断AIS尤其是CAIS, 需要通过临床表现、家族史、激素水平测定、核型分析、AR基因突变检测等各项指标综合判断[10], 并且需要与Swyer综合征[16]、类固醇5α 还原酶2缺乏症(steroid 5α -reductase deficiency, 5ARD)[17]、永久性苗勒氏管综合征(persistent Mü llerian duct syndrome, PMDS)[18]以及17β 羟类固醇脱氢酶(hydroxysteroid dehydrogenase, HSD)缺乏症[19]等相鉴别。明确诊断AIS需要基因检测, 由于经济及技术原因, 本课题组并未进行这部分实验, 但本文中患者的典型临床症状、影像学及化验室指标足以明确CAIS诊断并指导下一步诊疗计划。
在管理AIS患者方面, 多学科联合专家组是目前最主流的治疗策略[7], 其中包括妇科、泌尿外科、内分泌科、精神科医师, 同时也需要遗传学以及病理解剖学的专家共同参与制定治疗方案[20]。近些年来, 对CAIS患者行睾丸切除术并在术后予激素替代治疗, 已逐渐被大家认可并广泛应用于临床实践中。同时, 对于有特殊需求的患者还可以行阴道成形术或人工阴道, 不仅从身体更从心理上恢复患者的健康, 提高患者的生活质量[5]。
睾丸切除术最主要的目的是预防睾丸癌变, 但手术时机临床并未统一[5], 在青春期阶段, 睾丸分泌睾酮并部分转化为雌激素从而维持女性表型, 不宜切除睾丸。另外, 近年研究表明, 对于CAIS患者, 出生后1年内生殖细胞数量急剧减少, 睾丸组织中未成熟生精小管含有极少量甚至没有生殖细胞, 因此青春期发生癌变风险极低(CAIS患者约为0.8%), 但此后睾丸组织恶变概率显著增加, 所以推荐在青春期结束或成年后早期进行手术[1, 3, 21, 22]。
激素替代治疗同样是一个不可或缺的环节, 持续口服雌激素并缓慢加量是目前的主要治疗方案, 主要通过外源性补充雌激素维持女性生理需要[11]。有研究发现, 睾丸切除术后促性腺激素水平显著提升, 雌激素替代治疗的负反馈作用仅能部分抑制促性腺激素的过度分泌, 而睾酮替代治疗则能够进一步稳定激素水平, 对于传统替代治疗效果不佳的患者是一种可选择的治疗手段[23, 24], 但此方法对CAIS患者的治疗效果及预后无改善作用。除手术及药物治疗外, 对患者性生活满意度的评估也是很重要的, 因其对患者的自尊心及自我认知甚至婚姻及生活有着密不可分的联系[25]。
从症状、体征、化验指标、影像学检查以及核型分析等综合考量, 本文患者符合CAIS特点且比较典型, 诊断较为明确。患者社会性别为女性, 因此治疗以雌激素替代治疗为主, 尽可能稳定其性激素水平, 避免雄激素过高引发的一系列症状及体征。同时, 在此后进一步随诊过程中, 也应尽可能地对患者进行循序渐进的心理辅导, 增强其对自身性别的认可程度, 改善心理状态, 提高患者的生活质量。
对患者家族史进行仔细询问后, 发现其大姐已婚已育, 无闭经, 其二姐及两位姨妈均已婚未育且原发性闭经, 无阴毛、腋毛发育, 因此考虑二姐、两位姨妈有AIS可能。但3位亲属除上述症状和体征外, 无其他相关检查结果, 具体情况未知, 暂且不能明确本病诊断, 因此尚不能明确是否为家族遗传性疾病。但若按照3位亲属皆患病的情况分析, 符合X连锁隐性遗传特点, 与本病相同(图6)。
 | 图6 系谱图Figure 6 Pedigree analysis |
术后第6天、第11天检测性激素水平, 两次指标分别为:FSH 75.75 IU/L和82.00 IU/L、LH 38.25 IU/L和42.20 IU/L、睾酮1.28 nmol/L和1.00 nmol/L、孕酮0.33 μ g/L和0.62 μ g/L、雌二醇20.44 ng/L和31.97 ng/L。FSH及LH与术前(FSH 68.14 IU/L、LH 31.67 IU/L)相比有明显升高, 主要原因是切除睾丸组织后雄激素合成功能骤降, 血液中含量逐渐减少, 通过负反馈作用刺激促性腺激素分泌增多。睾酮水平较术前(4.13 nmol/L)显著改善, 表明睾丸组织切除较为完整, 提示良好预后。雌二醇及孕酮与术前相比(29.08 ng/L、0.73 μ g/L)先下降后升高, 考虑其下降原因为切除睾丸后, 雄激素代谢途径中的孕酮及雌二醇失去合成来源。而由于患者已无靶器官接受FSH及LH的反馈调节, 因此考虑二者升高可能是由于性腺外(如肾上腺皮质网状带、脂肪细胞等)性激素分泌功能的持续存在或继发增强, 导致雌二醇和孕酮的激素水平可有轻度升高或上下浮动, 但两种激素在3次检测中均处于绝经期女性激素水平, 波动范围不大, 对治疗及预后没有显著的参考意义。后续的雌激素替代治疗将进一步改善并稳定上述激素的水平。
该患者的特殊之处在于其最初以反复腹水及间断腹痛为主诉就诊, 术中发现膀胱顶壁瘘, 明确该瘘为腹水成因。因此, 对膀胱瘘的成因, 提出以下3种假说:
第1种假说为直接手术损伤。患者8年前曾因膀胱癌行TURBt及术后灌注化疗, 术中可能伤及膀胱顶壁, 造成局部膀胱组织损伤, 进而形成溃疡、穿孔, 最终演变为膀胱瘘。但患者原发肿瘤位于侧壁, 术中损伤顶壁可能性较小, 且如果为手术损伤, 腹水及间断腹痛等症状会在术后不久出现, 但患者术后3年余才出现上述症状。即便如此, 也不能排除术中损伤较表浅而术后缓慢形成瘘管的可能性。
第2种假说为癌症局部复发。患者8年前曾患膀胱癌, 且此次术后病理提示瘘管周围膀胱组织呈灶状尿路上皮非典型增生, 故怀疑癌症局部复发, 肿瘤浸润性生长, 导致局部组织坏死穿孔, 进而突破肌层、浆膜层, 最终穿透膀胱顶壁形成瘘管。但此次病理仅提示非典型增生, 并未发现明确浸润性生长表现, 且原发肿瘤为低级别尿路上皮癌, 复发且突破浆膜层可能性不大, 但不能除外。
第3种假说为脐尿管发育异常。术中发现膀胱顶壁瘘的位置位于脐正中韧带与膀胱的连接处, 因此考虑脐尿管发育异常的可能。胚胎发育第5周时, 泄殖腔分化为背侧的肛直肠管和腹侧的尿生殖窦, 后者的上段分化为膀胱, 其上连接尿囊, 尿囊进而退化为脐尿管, 成为连膀胱顶向脐延伸的管状通道, 位于腹横筋膜和腹膜之间的Retzius间隙。正常情况下, 在胚胎发育的第4、5个月时, 随着膀胱下降进入盆腔, 脐尿管逐渐萎缩闭合, 退化为无功能的纤维化结构, 即脐中韧带[26, 27]。但若未完全闭锁, 主要分为以下4种类型:(1)脐尿管未闭:管腔完全未闭合; (2)脐尿管窦道:仅脐部未闭合; (3)脐尿管囊肿:两端闭合而中部管腔未闭合; (4)脐尿管憩室:与膀胱连接处未闭合。依据本例患者瘘的位置, 考虑为脐尿管憩室继发破裂的可能性大。脐尿管憩室一般无明显症状, 但由于憩室的特殊解剖位置及结构, 易引起尿液反流、杂质沉积, 反复刺激从而继发憩室内炎症、感染、结石、非典型增生, 甚至引起膀胱排尿功能障碍, 导致憩室内压力进一步增加, 使解剖结构薄弱的憩室壁更容易产生破裂穿孔[28]。Chilakala等[29]报道1例先天性脐尿管憩室破裂引发尿性腹水的案例, Kumar等[30]报道1例腹腔镜阑尾切除术损伤脐尿管憩室导致穿孔引发腹水的案例, 说明脐尿管憩室由于自身因素或手术损伤均较易发生破裂穿孔。考虑到本例患者发病年龄大、病程不长且影像学未提示相关表现, 先天性脐尿管憩室自发出现穿孔的可能性不大, 并且憩室较小, 影像学不易发现, 因此, 怀疑可能为8年前TURBt术中对膀胱的过度牵拉或局部损伤刺激影响憩室结构稳定性或引起较轻的破损, 手术后随着膀胱排尿、储尿及收缩扩张功能的恢复, 尿液、沉积物等对憩室壁产生持续炎症性或机械性刺激, 进一步加重憩室壁的破坏, 最终形成膀胱瘘, 这也符合TURBt和出现症状之间的时间差。但目前尚无研究及个案报道支持或提示脐尿管发育及退化过程与生殖管道的胚胎发育过程有明确相关性。AR基因突变对两者是否均有影响以及后者的异常发育是否会对前者的退化造成影响, 两者是具有相关性还是仅仅为合并关系, 还需要进一步的研究。
The authors have declared that no competing interests exist.
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|