{kind=link}
{kind=link}
甲基丙二酸尿症相关肺高血压临床特点与基因突变
[刘雪芹1  , 闫辉
, 闫辉1 , 邱建星2 , 张春雨1 , 齐建光1 , 张欣1 , 肖慧捷1 , 杨艳玲1 , 陈永红1 , 杜军保1 ]
, 闫辉|
|


目的 总结15例以肺高血压(pulmonary hypertension, PH)为突出表现的甲基丙二酸尿症(methylmalonic aciduria, MMA)患儿临床特点及基因检测结果,提高对甲基丙二酸尿症相关PH临床表现的认识及诊治水平。方法 回顾性分析2012年5月至2016年5月北京大学第一医院儿科诊断治疗的15例以PH为突出表现的MMA患儿临床特点、诊断治疗经过、基因突变分析及随访结果,MMA的诊断标准为尿中甲基丙二酸水平>正常值的100倍,检测血浆总同型半胱氨酸(homocysteine,Hcy)和脑利钠肽(brain natriuretic peptide,BNP)水平。PH的诊断标准采用多普勒超声经三尖瓣反流估测的肺动脉收缩压(pulmonary arterial systolic pressure, PASP)>40 mmHg(1 mmHg=0.133 kPa)。结果 (1)起病特点:15例患儿中男10例,女5例,年龄0.5~13.8岁,平均(5.0±4.3)岁,PH起病年龄(3.7±3.5)岁,其中早发型5例,晚发型10例,10例PH症状为MMA首发表现,5例在MMA起病后3~72个月出现PH症状。(2)临床表现:气促和/或呼吸困难11例,口唇发绀11例, 乏力和/或活动耐力下降6例,水肿4例;PH国际卫生组织功能分级(WHO FC)为Ⅱ级4例,Ⅲ级5例,Ⅵ级6例,平均(3.1±0.8)级。(3)多系统损害:肾损害14例,表现为血尿及蛋白尿,5例为慢性肾脏病(chronic kidney disease,CKD),8例大细胞性贫血,4例伴轻-中度智力运动发育落后,5例亚临床甲状腺功能减低。(4)辅助检查:15例患儿经超声心动图三尖瓣反流测量的肺动脉收缩压49~135 mmHg,平均(90.3±23.9) mmHg;血浆总Hcy显著升高[35.0~221.0 μmol/L,平均(121.2±48.2) μmol/L],其中11例>100 μmol/L; 12例血BNP水平不同程度升高[21.0~4995.0 ng/L,中位值794 ng/L,其中12例>300 ng/L],血气分析发现存在不同程度低氧血症,动脉血氧饱和度平均81.4%±8.4%(70%~94%)。(5)肺部高分辨CT(high resolusion CT, HRCT):9例小叶中心磨玻璃密度结节及肺小叶间隔增厚,提示肺静脉闭塞病(pulmonary veno-occlusive disease, POVD),其中3例伴肺部炎症或肺水肿,另3例伴弥漫间质浸润呈网格样改变,提示肺间质病变。(6)基因检测结果:10例均为MMACHC基因复合杂合突变(cblC型),共发现5种已报道突变,其中10例c.80A>G突变,6例同时存在c.609G>A突变。(7)治疗及随访结果:所有患儿均给予羟钴胺肌肉注射及甜菜碱等治疗,11例给予肺动脉高压靶向药物,住院治疗后PASP、血Hcy、BNP均显著降低,2例死亡,13例随访11~64个月,平均(27.5±19.0)个月,临床症状均缓解,除1例外,肺动脉压力均在3~6个月恢复至正常,随访PH无复发,多系统损害明显恢复。结论 PH是MMA合并型的严重并发症,多发生于晚发型男性患儿,临床以气促、呼吸困难和发绀症状为突出表现,多存在低氧血症,HRCT多呈POVD改变,PH常与肾受累同时存在,及时诊断并给予针对MMA的治疗以及恰当抗PH治疗,肺动脉压力短期内多可恢复正常,重症患儿可危及生命。MMACHC基因c.80A>G可能是MMA相关PH的热点突变。
, YAN HuiObjective: To deepen our understanding of Methylmalonic aciduria (MMA) associated pulmonary hypertension (PH) by analyzing the characteristics of clinical presentation, pulmonary high resolusion CT(HRCT), treatment response and gene mutation.Methods: This study includes 15 cases of pediatric patients with MMA associated PH diagnosed and treated in Peking University First Hospital pediatric department between May 2012 and May 2016 with symptoms of PH as their leading presentation. Clinical symptoms and signs were recorded, Routine blood laboratory examinations was done including arterial blood gas analysis. Plasma total homocysteine (Hcy) and brain natriuretic peptide(BNP) level were measured. MMA gene mutation was analyzed. Chest HRCT was done in most of the patients. Standard treatment strategy to MMA and PH was given and follow up study was done, and the related literature was reviewed. Statistical analysis was done. The diagnosis of MMA was made by methylmalonic acid level >100 times the normal value in the urine. The diagnosis of PH was made by pulmonary arterial systolic pressure (PASP)>40 mmHg, which was estimated by the measurement of tricuspid regurgitation velocity through Doppler Echocardiography.Results: (1) Patient characteristics: There were 10 male and 5 female patients diagnosed as MMA associated PH, aged 0.5 to 13.8 years, with an average of (5.0±4.3) years. The age of onset of PH was (3.7±3.5) years, with an early onset type MMA in 5 cases and late-onset type in 10 cases. (2) Clinical presentation: Among the 15 cases of MMA, the first symptoms were associated with PH in 10 cases , so PH and MMA were diagnosed at the same time, and PH was diagnosed 3 to 72 months post MMA presentation in the other 5 cases. The main presentations of PH were techypnea /dyspnea and cyanosis in 11 cases each, weakness and fatigue on exertion in 6 cases, and edema in 4 cases. PH WHO functional classification (WHO FC) was ClassⅡin 4 , Class Ⅲ in 5 and Class Ⅵ in 6 cases, with an average of Class 3.1±0.8. Multi-system involvements were common with the highest frequency in the kidney (14 cases). Macrocytic anemia was present in 8 cases and sub-clinical hypothyroidism in 5 cases, and mild to moderate mental retardation in 4 cases. (3) Laboratory examination: PASP of the 15 patients was from 49 to 135 mmHg, with an average of (90.3±23.9) mmHg. Total blood Hcy level was severely elevated to (121.2±48.2) μmol/L (range: 35.0-221.0 μmol/L), and Hcy >100 μmol/L within 11 cases. Plasma BNP level was also elevated, median 794 ng/L (range: 21.0-4 995.0 ng/L) with 12 cases >300 ng/L. Blood gas analysis showed low arterial blood oxygen saturation between 70% and 94%, with an average of 81.4%±8.4%. (4) Chest HRCT: chest HRCT showed a diffuse ground-glass centrilobular nodular opacities with septal line thickening in the lungs in 9 cases, and with associated mediastinal lymph node enlargement in 1 case, which indicated pulmonary veno-occlusive disease (PVOD), a rare type of pulmonary arterial hypertension (PAH). There was lung infection or edema in 3 cases , and interstitial infiltration and mesh-like feature in other 3 cases, which was inferred to interstitial lung disease. (5) Gene mutation: Genetic testing was done in 10 cases, totally 5 reported disease-causing mutations were found. There were 100% presence of MMACHC c.80A>G mutation in all the 10 patients tested, with the allelic genes of c.609G>A mutation in 6 patients, including a sister and a brother from the same parents. (6). Treatment and follow up: Intramuscular hydroxocobalamin or vitamin B12 was given to all of the patients, together with betaine, levocarnidtine, folinic acid and vitamin B6. According to the severity of PH, single or combined PAH targeted drugs was given to 11 cases. By an average of (20.0±13.5) days of in-hospital treatment in 13 patients (excepting 1 case treated as outpatient), symptoms remarkably resolved, WHO FC reduced to an average of Class 2.4±0.9, PASP dropped to (69.4±21.3) mmHg, and plasma Hcy and BNP level were decreased to (74.9±25.9) μmol/L and (341.6±180.2) ng/L, respectively. The above values all reached statistical significance ( P<0.05) compared with each related value before treatment. There were 2 patients who expired during hospitalization despite of treatment. At the end of 3 months’ follow up, all of the 13 patients disposed oxygen, and PASP significantly dropped to 38.7±7.9 mmHg, and plasma BNP returned to normal, but plasma Hcy level showed no further decline. At the last follow up of 27.5±19.0 (range: 11-64) months, all the patients’ PASP remained normal except for the 13.8-year-old boy with 6 years-long history of MMA and almost 3.6 years’ history of PH still having PASP 58 mmHg.Conclusion: PH is a severe complication of MMA combined type, especially cblC type, it is more often happens in late-onset type of male patients and can be the first and leading manifestations of MMA. Its clinical symptoms are urgent and severe, characterized by tachypnea/dyspnea and cyanosis, and sometimes right heart failure, hypoxemia is usually present, chest HRCT is often indicative of PVOD, lung edema and interstitial lung disease may occur. Rapid diagnosis and targeted treatment of MMA with appropriate anti-PAH medication can reverse PH and save life. MMACHC gene c.80A>G mutation may be the hot point of MMA cblC type associated PH.
甲基丙二酸尿症(methylmalonic aciduria, MMA)又名甲基丙二酸血症, 是先天性有机酸代谢病中最常见的疾病之一, 呈常染色体隐性遗传, 可分别由甲基丙二酰辅酶A变位酶或其辅酶钴胺素(维生素B12)代谢基因缺陷导致, 其中cb1C、cblD-MMA/HC、cblF及cblJ缺陷可同时导致腺钴胺及甲钴胺合成缺陷, 代谢改变呈现为MMA合并同型半胱氨酸血症, 又称MMA合并型, 其中以MMACHC基因突变的cblC型最为常见[1, 2]。MMA临床常出现神经、肾、血液等多器官系统受累, 心血管系统受累相对少见, 文献报道本病并发肺高血压(pulmonary hypertension, PH)常可造成患儿死亡[3, 4, 5], 北京大学第一医院儿科近4年诊治15例以PH为突出表现的MMA, 病情多危重, 及时诊断和治疗取得了较好疗效, 现对该组病例临床特点、诊断治疗过程、随访情况及基因突变进行回顾性分析。
1.1.1 研究对象
选择2012年5月至2016年5月在北京大学第一医院儿科诊断治疗及随访的以PH症状就诊的MMA患儿共15例进行回顾性分析。
1.1.2 甲基丙二酸尿症的诊断及分型
患儿尿液标本经色谱/质谱联用法进行尿有机酸分析(尿代谢筛查), 尿甲基丙二酸浓度升高> 正常值的100倍, 则临床诊断为MMA, 血液标本以液相色谱/质谱分析法进行血氨基酸及酰基肉碱谱分析(血代谢筛查), 若血蛋氨酸水平降低, 丙酰肉碱水平升高, 则支持诊断[1, 2]。血浆总同型半胱氨酸(homocysteine, Hcy)测定采用免疫荧光偏振法, 若测定值高于正常参考值高限(15 μ mol/L), 则诊断为高Hcy血症, 尿甲基丙二酸与血Hcy均升高则诊断为MMA合并型[1, 2]。根据MMA临床分型标准, 起病年龄≤ 1岁为早发型, > 1岁为晚发型[2]。
1.1.3 排除标准
所有患儿同时进行血维生素B12及叶酸水平测定, 排除维生素B12及叶酸缺乏病例, 同时也排除不伴有MMA的单纯性高Hcy血症。
1.1.4 肺高血压诊断标准
首先对患儿进行常规超声心动图检查排除右心室流出道及肺动脉狭窄病变, 采用经收缩期三尖瓣反流速度方法估测肺动脉收缩压(pulmonary arterial systolic pressure, PASP), PASP> 40 mmmHg(1 mmHg=0.133 kPa)诊断为PH[6]。
1.2.1 采集患儿临床资料
包括性别、年龄、身高、体重、首发症状及PH相关症状及其出现时间、其他系统受累表现。根据PH临床表现, 按国际卫生组织(WHO)PH功能分级诊断标准进行功能分级(WHO FC)[7, 8]。
1.2.2 对患儿进行下列辅助检查
(1)尿液及血液代谢筛查; (2)血浆总Hcy水平测定; (3)血浆脑利钠肽(brain natriuretic peptide, BNP)检测; (4)经胸常规超声心动图及测定三尖瓣反流速度并计算PASP, 测值取连续3个心动周期测量的平均值; (5)计算机断层扫描肺动脉造影(CTPA)及肺部高分辨CT(high resolusion CT, HRCT); (6)常规检验:血、尿、便常规, 血生化、血维生素B12及叶酸水平检测; (7)其他检查:动脉血气分析或经皮血氧饱和度(SaO2)测定、X线胸片、心电图、部分患儿进行了核素肺通气灌注扫描。以上各项检查中, (1)项在北京大学第一医院代谢筛查室进行, (2)~(7)项由北京大学第一医院相应辅助科室完成, 以上各项检验、检查均在入院后3日内进行, 必要时分别在出院前3日及随访时进行复查。另对患儿进行眼科检查, 了解有无视力下降及视网膜病变。
1.2.3 甲基丙二酸尿症基因学检测
基因检测与测序由北京德易东方转化医学研究中心完成, 提取患儿及其父母外周血全基因组DNA, 采用二代测序方法捕获目标基因全外显子, 一代测序(Sanger法)验证。基因检测与测序前患儿父母签署知情同意书。
1.2.4 治疗
临床确诊或高度疑诊MMA, 即给予羟钴胺或维生素B12 1 mg/d肌肉注射, 出院后改为隔日1 mg肌肉注射, 同时给予甜菜碱250 mg/(kg· d)口服, 另给予左旋肉碱、亚叶酸钙、维生素B6等辅助治疗药物。患儿入院后给予吸氧, 有心力衰竭者根据病情给予限液、利尿剂、地高辛、多巴胺等治疗, 根据WHO FC, 按PH治疗指南给予抗肺动脉高压(pulmonary arterial hypertension, PAH)靶向药物[7, 8]。
1.2.5 随访
患儿出院后持续给予上述针对MMA的药物治疗, 每3~6个月来北京大学第一医院门诊或电话随访, 复查血浆总Hcy及BNP水平, 复查常规超声心动图, 复测PASP, 并调整治疗方案。
应用SPSS17.0软件进行统计学分析, 病例计数以例数(%)表示, 计量资料以均数± 标准差表示, 各项检测数据计量资料治疗前后及其与随访数据的比较采用配对t检验, P< 0.05认为差异有统计学意义。
2012年5月至2016年5月, 北京大学第一医院儿科诊治MMA相关PH共15例, 其中2例为同胞姐弟, 均为MMA合并高Hcy血症, 即MMA合并型。15例中男10例, 女5例, 男女之比为2 ∶ 1, 年龄0.5个月至13.8岁, 平均(5.0± 4.3)岁(中位值 3.9岁), MMA起病年龄(3.7± 3.5)岁(中位值2.6岁), 其中早发型5例(33.3%), 晚发型10例(66.7%), 发生PH年龄(4.7± 3.9)岁(中位值3.8岁), 14例患儿住院治疗, 1例门诊治疗, 14例住院时间6~46 d, 平均 (20.0± 13.5)d(中位时间17.5 d), 2例危重患儿(病例13、病例14)分别于住院第7天及第10天死亡, 余13例存活。
2.2.1 临床表现特点
2.2.1.1 起病特点 15例患儿均以PH症状为主诉就诊, 其中10例(66.7%)PH为首发临床表现, PH与MMA同时确诊, 其余5例(33.3%)(病例3、4、6、10、12)在其他系统受累之后出现PH症状, MMA起病至诊断PH间隔3~72个月, 平均为(40.2± 30.8)个月, 此5例患儿起病分别表现为智力运动发育落后、心脏扩大、贫血、血尿、蛋白尿及慢性肾脏病(chronic kidney disease, CKD, 表1), 其中病例3和病例10分别在8月龄及3月龄于院外确诊MMA, 但未规范治疗。
2.2.1.2 症状特点 15例患儿中主诉气促和/或呼吸困难11例(73.3%), 口唇和/或肢端发绀11例(73.3%), 乏力和/或活动耐力下降6例(40.0%), 水肿4例(26.7%, 表1)。
| 表1 15例甲基丙二酸尿症相关PH患儿临床表现 Table 1 Clinical presentations of 15 cases of MMA associated PH |
2.2.1.3 体征特点 患儿均有不同程度呼吸急促, 呼吸空气氧状态下, 8例明显口唇发绀, 3例心前区饱满, 4例心界扩大, 12例肺动脉瓣听诊区第2心音亢进, 3例闻及3/6级三尖瓣反流收缩期杂音, 4例颜面或双下肢水肿, 4例杵状指, 4例存在高血压。
2.2.1.4 WHO FC Ⅱ 级4例, Ⅲ 级5例, Ⅵ 级6例, 平均(3.1± 0.8)级。
2.2.1.5 其他系统受累情况 (1)肾:共14例(93.3%)存在肾受累, 包括3例镜下血尿, 11例镜下血尿和蛋白尿, 共5例(33.3%)诊断为CKD; (2)血液系统:8例(53.3%)存在大细胞性贫血; (3)神经系统:4例(26.7%)智力运动发育落后, 轻、中度各2例, 其中3例为早发型; (4)其他系统:5例(33.3%)存在亚临床甲状腺功能减低症, 另1例水平眼震, 12例行眼科会诊均未报告视网膜病变及其他眼科异常(表1); (5)血生化检查结果:4例轻度代谢性酸中毒, 4例肝酶升高, 9例尿素氮升高, 3例肌酐升高。
2.2.2 辅助检查
2.2.2.1 肺动脉收缩压 15例患儿入院经三尖瓣反流估测的PASP为49~135 mmHg, 平均(90.3± 23.9) mmHg, 均为中-重度PH(表2)。
| 表2 15例甲基丙二酸尿症相关PH患儿入院治疗前后各指标的变化及比较 Table 2 Parameters changes after in-hospital treatment in 15 patients |
2.2.2.2 血浆同型半胱氨酸水平 15例(100%)血浆总Hcy治疗前均显著升高, 平均(121.2± 48.2) μ mol/L (35.0~221.0 μ mol/L), 其中11例> 100 μ mol/L, 为重度升高, 4例中度升高(表2)。
2.2.2.3 血维生素B12及叶酸水平 15例均在正常范围。
2.2.2.4 血浆BNP水平 12例(80%)不同程度升高, 平均(1 652.3± 1 866.7) ng/L(21~4 995 ng/L), 中位值794 ng/L, 其中12例> 300 ng/L, 3例超出测值高限, 余3例< 100 ng/L(表2)。
2.2.2.5 血气分析 15例入院时均存在不同程度低氧血症, SaO2为81.4%± 8.4%(70%~92%, 表2)。
2.2.2.6 心电图 10例(80%)右心室肥大或右心室高电压, 3例双心室肥大, 4例ST-T改变。
2.2.2.7 超声心动图 11例(73.3%)存在右心室肥厚或/及扩大, 10例同时存在右心房扩大, 2例伴左心室扩大, 左心室收缩功能均正常, 15例均存在轻至中度三尖瓣反流, 6例同时存在轻度肺动脉瓣反流, 另2例存在小型室间隔缺损。
2.2.2.8 X线胸片 15例中8例表现为肺纹理增粗模糊, 8例伴有肺部渗出, 影像诊断为支气管炎及肺炎, 3例肺水肿, 2例肺间质病变, 2例报告正常, 病例11胸片可见肺动脉段凸出, 肺纹理增多, 双下肺野有增厚的小叶间隔线(图1)。
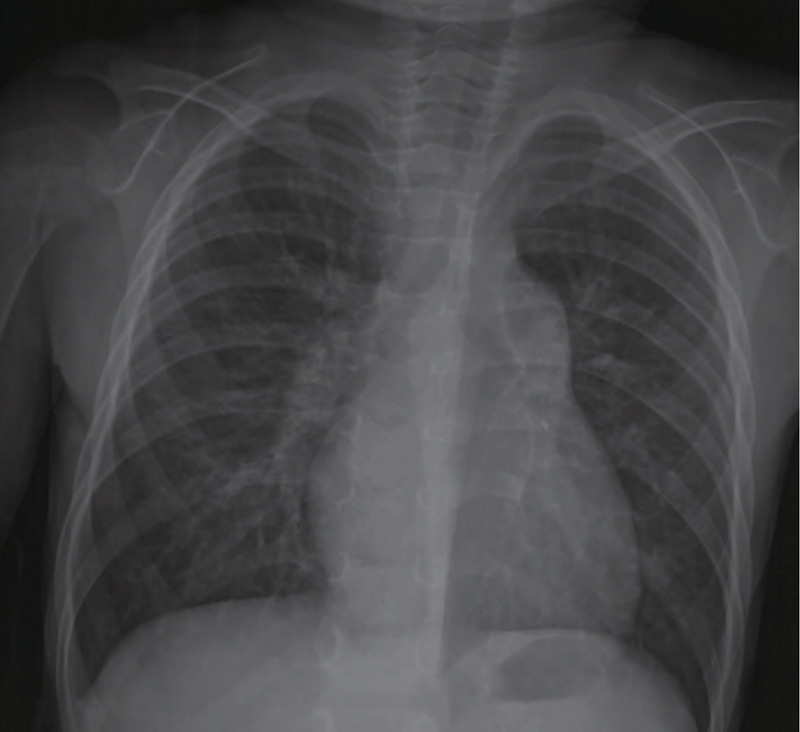 | 图1 病例11 的X线胸片Figure 1 Chest X-ray film of case 11 |
2.2.2.9 CTPA及HRCT 13例患儿进行了此两项检查, CTPA示全部患儿存在不同程度主肺动脉扩张及右心室饱满或扩大, 2例少量心包积液。HRCT示9例(69.2%)肺小叶中心磨玻璃密度结节及肺小叶间隔增厚, 1例伴纵膈淋巴结肿大, 提示肺静脉闭塞病(pulmonary veno-occlusive disease, PVOD), 其中3例(23.1%)伴肺部炎症或肺水肿, 另3例(23.1%)弥漫间质浸润呈网格样改变, 提示肺间质病变, 余1例血管束增粗、肺小动脉迂曲, 提示肺血管病变。13例均除外肺血管畸形。病例1肺部HRCT示PVOD改变可见小叶中心磨玻璃样结节及小叶间隔增厚(图2)。
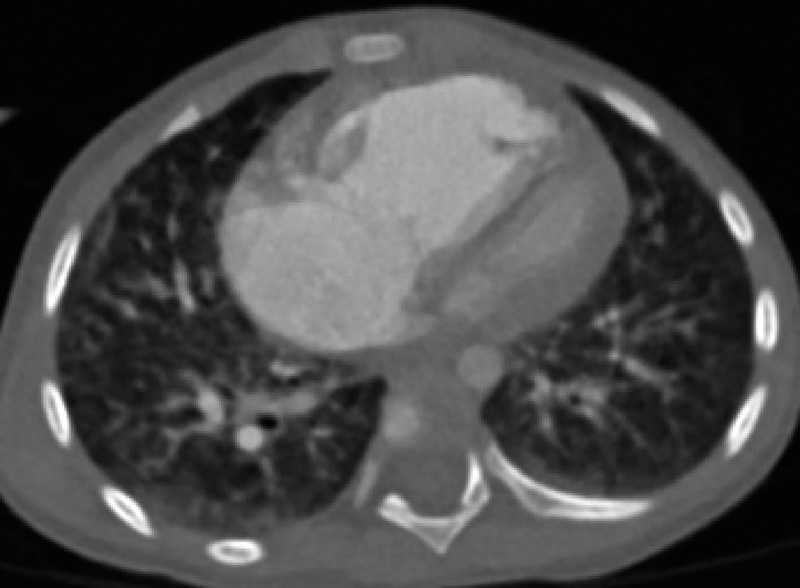 | 图2 病例1的胸部HRCTFigure 2 Chest high resolusion CT of case 1 |
2.2.3 基因检测结果
10例患儿行MMA基因组基因测序检测, 结果均为MMACHC基因复合杂合突变, 即cblC型, 共发现5种突变, 均为已报道致病突变, 10例(100%)均存在c.80A> G突变, 包括2例同胞姐弟在内的6例同时存在等位基因c.609G> A突变, 基因型为c.80A> G/c.609G> A, 另2例为c.656_658delAGA缺失突变, 余2例分别为c.658_660delAAG及c.331G> T突变, 经家系验证, 各突变均遗传自患儿生物学父母, 基因检测结果见表3。
| 表3 甲基丙二酸尿症相关PH基因测序结果 Table 3 Gene testing results in 10 patients |
患儿入院后确诊或高度怀疑MMA, 即给予针对MMA的标准治疗, 包括肌肉注射羟钴胺或维生素B12及口服甜菜碱等药物, 全部患儿给予吸氧, 10例给予利尿剂、地高辛抗心力衰竭治疗, 其中4例静脉应用多巴胺、米力农; 11例(73.3%)给予PAH靶向药物, 其中8例单药(西地那非3例、他达拉非2例、波生坦3例), 3例(病例1、6、11)两药联合(波生坦加他达拉非、安立生坦加他达拉非、波生坦加伐地那非), 其余4例未用靶向药物(病例3、5、9、14)。2例危重患儿(病例13、14)在治疗过程中分别于住院第7天及第10天死亡, 死因为肺动脉高压危象及严重心力衰竭; 其余12例患儿住院期间呼吸困难均明显缓解、水肿均消失, 吸氧流量降低, 出院前复查PASP显著下降, 血Hcy水平、血BNP均显著降低, WHO FC显著降低, 提示临床好转, 住院治疗前后各指标变化见表2。
对12例出院患儿及1例(病例9)门诊患儿随访11~64个月, 平均(27.5± 19.0)个月。出院后3个月首次复诊, 9例复诊患儿气促及呼吸困难均完全缓解, 呼吸室内空气经皮血氧饱和度达93%~97%, 3例需间断吸氧, 超声心动图复测PASP, 6例正常, 余3例较出院时明显降低, 最高为50 mmHg(病例6), 于出院后6个月随访的13例中仅病例6仍存在中度PAH, 余12例PASP均在正常范围, 包括未用靶向药物治疗的3例。至末次随访, 仅病例6仍未恢复正常。经统计学分析, 出院3个月随访, PASP较住院治疗后进一步降低(38.7± 7.9 vs. 69.4± 21.3, t=6.485, P< 0.01)。3~6个月随访, 血BNP均降至正常、血Hcy水平较出院时无明显变化, 仍多处于中度升高水平, 2例CKD患儿(病例6和12)保持高值。10例应用PAH靶向药物的存活患儿中, 9例于随访3个月停用, 仅病例6 诊断PH已3.6年, 随访19个月PASP无进行性降低, WHO FC为Ⅱ 级, 持续波生坦治疗。多系统损害情况:出院后3~6个月随访, 除病例6及病例12外, 肾受累均好转, 8例血尿和/或蛋白尿消失, 12例贫血均恢复, 4例智力运动发育落后患儿发育明显进步, 5例亚临床甲状腺功能减退患儿甲状腺功能均恢复正常, 各次随访各指标变化见表4。
| 表4 13例甲基丙二酸尿症相关PH患儿各指标随访结果 Table 4 Parameters during followup in 13 patients |
本研究通过对最近4年来本院诊断的15例MMA相关PH研究发现, 患儿均为MMA合并型, PH多发生于晚发型男性患儿, 起病年龄跨度大, 主要发生在学龄前阶段, 多为中-重度PH, 而且在多数患儿为MMA首发表现, 少数发生在其他系统受累之后, 临床上以气促/呼吸困难、发绀等缺氧表现突出, 可伴有乏力、水肿等心力衰竭症状, 同时多并存肾损害及大细胞性贫血。患儿确诊时均存在不同程度低氧血症, 血BNP不同程度升高; 肺部HRCT具有一定特征性, 多呈PVOD表现。本病及时诊断及正确治疗, 绝大多数患儿短期内代谢改善, 症状好转、低氧血症消失, 肺动脉压力、血BNP于治疗后3~6个月多可恢复正常, 且持续治疗原发病, PH无反复, 但晚期危重者可短期内死亡。
应用多普勒超声心动图经三尖瓣反流估测PASP是临床筛查诊断PH的传统方法[6], 本组患儿经三尖瓣反流估测的PASP值均在40 mmHg以上, 故PH诊断明确, 且患儿PASP较高, 均为中-重度, 平均值达90 mmHg, 最高135 mmHg, 提示肺血管病变较严重。本组患儿WHO FC平均为(3.1± 0.8)级, Ⅲ ~Ⅳ 级者占73.3%, 4例存在右心衰竭临床表现, 12例血BNP水平> 300 ng/L, 3例显著升高超过可测值高限, 提示多数患儿病情已达高危状态[7, 8]。按目前肺高血压最新临床分类, MMA并发PH属第5类[7], 为多种机制导致。根据文献报道病例心导管检查结果, 提示本病相关PH为毛细血管前PH[3, 4, 5, 9, 10, 11], 即PAH, 文献曾报道3例死亡患儿肺组织病理结果为肺血管病及PVOD[3, 5], 亦支持毛细血管前PH诊断。虽然目前PH的确诊以右心导管测量的肺动脉平均压≥ 25 mmHg为金标准[7], 诊断PAH同时应具备肺小动脉嵌顿压< 15 mmHg, 但鉴于本组患儿起病时病情多危重, 且文献报道本病患儿行心导管检查后可能出现PH危象或心力衰竭加重而死亡[4, 5], 加之本组患儿病因明确, 治疗效果较好, 考虑到风险效益比, 故本研究中未行右心导管检查。
本组15例患儿均伴有高Hcy血症, 与文献报道一致[3, 4, 5, 9, 10, 11, 12], 提示MMA相关PH多发生于其合并型。本组患儿以男性晚发型患儿为主, 占2/3病例, PH起病年龄分布广泛, 多在学龄前期发病, 仅5例为早发型婴儿期起病, 结合迄今文献报道的13例, 发病年龄0.2~18.0岁, 也仅有两例为早发型, 其中性别信息明确的10例, 7例为男性[3, 4, 5, 9, 10, 11, 12], 故提示MMA相关PH可能晚发型男性患儿易患, 此特点值得重视。
本组患儿中10例以PH为MMA首发临床表现, 5例在出现其他系统表现之后3~72个月发生PH, 起病较急, 病情较危重, 多以气促/呼吸困难、发绀等缺氧症状为突出表现, 存在不同程度低氧血症、重症病例出现右心衰竭, 2例患儿在入院治疗过程中死亡, 前述国外报道的13例, 临床表现与本组患儿相似, 其中7例死亡[3, 4, 5, 9, 10, 11, 12], 提示本病危重患儿存在死亡高风险。呼吸困难、发绀等缺氧症状多为儿童特发性PAH及先天性心脏病相关PAH的晚期临床表现[13], 而MMA相关PH患儿起病即出现上述危重症状, 考虑与PVOD导致气体弥散障碍有关[14], 此特点可作为本病早期诊断线索。基于上述临床特点, 在对MMA患儿的诊治中, 有必要对MMA合并型尤其晚发型男性患儿进行超声心动图筛查, 有助于早期发现PH; 另外, 对于起病急、存在缺氧症状的PH患儿, 需要进行代谢筛查及血Hcy检测, 以明确有无MMA。
本组15例患儿均存在多系统损害, 尤以肾受累最为多见, 除一例早发型(病例14)外, 14例均存在肾损害, 表现为镜下血尿和蛋白尿, 其中9例为本次住院后检查首次发现, 5例符合CKD, 3例年长儿(病例6、10、12)分别在肾受累4年、6年及5年后出现PH。研究已表明, MMA肾受累病理特征为血栓性微血管病, 临床可表现为不典型溶血尿毒综合征[15], Kö mhoff等[3]曾报道5例肾受累患儿同时存在PH, 其中3例死于右心衰竭, 死亡病例尸解存在肺血管病变及PVOD。本组资料结合文献, 提示MMA患者肺小血管与肾微血管可同时受累, 肾损害与PH可同时或先后出现, 但由于PH早期无症状且症状隐匿, 临床表现可能为晚于肾损害出现的血尿/蛋白尿, 因此, MMA合并型患儿若尿常规检查结果异常, 应及时进行心脏超声筛查, 了解有无PH。
本组1/3患儿存在亚临床甲状腺功能减低症, 考虑可能与MMA导致的腺体细胞能量代谢障碍有关。另外, 本组PH患儿血液系统受累亦较常见, 表现为大细胞性贫血, 对MMA诊断有提示作用。早发型MMA患儿神经系统受累常见且突出[1, 2], 本组仅4例轻-中度发育落后, 3例为早发型, 未发现文献报道常见的视网膜病变, 可能与本组患儿主要为晚发型有关。
本组患儿肺部HRCT表现较有特征性, 9例存在肺小叶中心磨玻璃结节伴小叶间隔增厚等PVOD影像学特点[14], 文献也有类似报道[3, 5, 11], 关于该种影像学表现的组织病理学变化, 在对死亡病例的研究中得以明确, Kö mhoff等[3]对3例死亡患儿的肺组织病理研究中发现2例为PVOD, 其与以肺小动脉重塑为特征的肺血管病变同时构成PH病理改变, Bouts等[5]也曾报道1例MMA相关PH死亡患儿存在PVOD。传统认为PVOD是一类病因及发病机制复杂的少见PH, 临床治疗困难, 预后较差[7, 14], 其主要病理特点为肺小静脉内膜纤维增生, 造成管腔狭窄或闭塞导致间质性肺水肿, 因气体弥散障碍造成低氧血症, 确诊依靠肺组织病理检查, 但由于肺活检存在较大风险, 而其HRCT影像学又具备一定特点, 故目前国内外临床诊断主要依靠HRCT, 其影像特征为两肺广泛分布的小叶中心磨玻璃结节、小叶间隔增厚, 可同时存在纵膈淋巴结肿大, 具备以上2~3项即支持诊断[14], 本组9例符合上述PVOD影像学特点, 提示PVOD可能为MMA相关PH的主要病理改变。除PVOD外, 由于患儿PASP恢复较快, 推测肺血管痉挛因素也可能参与了本病PH的发生。另外, HRCT提示存在的肺泡渗出、肺水肿、肺间质病变等, 也可能参与本病低氧血症及PH的发生。Brandstetter等[16]曾对1例本病死亡婴儿进行肺组织病理检查, 发现存在肺充血、肺水肿、肺透明膜形成以及肺小动脉血栓形成, 由此可知, MMA相关PH的形成可能为多种病理机制参与, 不同患者不同病期导致PH的主要病变可能存在差异, 肺部影像学随之表现出多样性, 而在病程较长、肺血管病变较严重者, 肺血管重塑可能难以逆转, 最终PH难以治愈, 本组病例6即不除外此种可能。
本组10例进行MMA基因组测序, 结果均为cblC型, 符合MMA以cblC型为主的流行病学特点[1, 2]。Liu等[17]对我国79例MMA cblC型患者基因分析发现, c.609G> A及c.80A> G突变位列该组患儿常见基因突变的第1位及第5位, Wang等[18]也曾报道c.609G> A是我国汉族患儿晚发型MMA cblC型最常见热点突变, 本组结果与此两项研究结论有所不同, 本组10例全部存在c.80A> G突变, 包括一对同胞姐弟在内的6例存在c.609G> A突变, 由此推测c.80A> G可能与MMA cblC型发生PH相关。迄今发现MMA共有7个基因型, 可引起MMA合并型者包括4个基因型, 即cblC、cblD-MMA/HC、cblF及cblJ[1, 2], MMA合并型的其他基因型造成与cblC型相似的代谢改变, 理论上也有并发PH可能, 但本组病例基因测序未发现其他基因突变, 可能与其他类型少见及本组病例样本量较小有关。
本组患儿入院后自确诊或高度疑诊即给予MMA针对性治疗药物, 主要为肠道外途径羟钴胺以及甜菜碱、左旋肉碱及亚叶酸钙等药物口服, 对重症患儿同时给予氧疗及抗心力衰竭治疗, 对于WHO FC Ⅲ ~Ⅳ 级患儿, 短期应用PAH靶向药物, 内皮素受体拮抗剂与磷酸二酯酶-5受体拮抗剂单药或联合应用, 取得满意疗效, 除两例危重患儿在入院后短期内死亡外, 其余13例PH症状均在住院期间至治疗后3~6个月缓解, 随访3个月多可停用靶向药物, 至末次随访已1~5年, 除病例6仍有中度PH, 其余12例PASP均维持正常, WHO FC 为Ⅰ ~Ⅱ 级, 对比文献报道13例中7例死亡[3, 4, 5, 9, 10, 11, 12, 16], 本组患儿治疗效果较好, 死亡率相对较低。值得指出的是, 本组3例未用靶向药物的存活患儿, PH也完全缓解, 提示原发病治疗的重要性, 而基础治疗和靶向药物可帮助重症患儿缓解心力衰竭, 度过危重阶段。本组中病例6为13.8岁年长儿, 总病程长达6年余, 诊断PH 3年余, 已随访1.5年, 持续应用波生坦, PASP仍为58 mmHg, 提示病程长、治疗晚的患儿, 可能肺血管病变较重, 虽代谢紊乱纠正, 肺血管重塑在一定程度上难以逆转。有研究认为cblC晚发型者[19], 除严重神经系统受累外, 血液、肾及外周血管受累多预后较好, 但PH是一类难以治愈的疾病[7, 8], MMA相关PH患儿远期疗效及预后如何, 尚需进一步观察研究。如前所述, MMA相关PH可为PVOD, 应用靶向药物有引起肺水肿可能, 但本组应用靶向药物的11例患儿, 均未出现肺水肿或原有肺水肿加重表现。需要特别指出的是, MMA是需要终生不间断治疗的代谢缺陷病, 治疗中断及感染等应激状态下, 患儿代谢紊乱可能加重, PH有出现反复的可能, 密切随防并及时调整治疗方案, 对于保持患儿代谢病及PH长期缓解有着非常重要的意义。本组MMA合并PH患儿均存在Hcy血症, 治疗前Hcy重度升高, 住院治疗后即显著下降, 但在长期随访中, 多数患儿血Hcy仍处于中等程度以上升高水平, 与文献报道大致相同[1, 15, 19], 表明目前针对MMA的治疗尚难以完全纠正本病代谢紊乱, 需探索更为积极的治疗方案, 残余代谢异常对肺循环血管的长期影响有待观察与研究。
甲基丙二酸尿症相关PH发生的确切病理生理机制尚未完全明确, 考虑首先与MMA的共同致病机制密切相关, 包括代谢产物的直接毒性作用、正常代谢产物丢失、甲基化能力下降、氧化应激等[1, 2], 研究已明确Hcy可损伤血管内皮并可导致血栓形成[20], 最近有研究发现甲基丙二酸可干扰血管内皮细胞线粒体产能[21], 造成内皮损伤, 此二者及其他毒性分子的共同作用以及基因功能的改变均可能是造成本病相关PH的分子机制, 而MMA相关PH与肾血栓性微血管病的同时存在, 提示PH与肾血管受累机制可能相似。本病尸解病例肺病理PVOD及以肺小动脉重塑为特征的肺血管病表现, 表明肺小动脉及小静脉均受累, 另外, 也仅有以肺小动脉重塑的病理表现者[3], 导致不同患者肺血管病变存在异质性的因素及其机制尚待进一步深入研究。
综上所述, PH是MMA合并型的严重并发症, 晚发型男性患儿多见, 多起病急, 以“ 气促/呼吸困难、发绀” 等缺氧症状为突出表现, 患儿多存在低氧血症, 重者发生心力衰竭, 患儿多同时或之前即出现肾损害、大细胞贫血, 而神经系统损害表现较轻, X线胸片表现多样, 肺部HRCT多呈PVOD及肺水肿、肺间质病变特征, 此临床特点需引起重视。晚期重症PH可迅速威胁患儿生命, 及时诊断和正确治疗原发病, 针对PH给予恰当的基础治疗及短期应用靶向药物疗效较好。MMA相关PH多发生于cblC型, MMACHC基因 c.80A> G可能为热点突变, 在对MMA合并型尤其cblC型患儿诊断治疗中, 应注意PH上述临床表现特点, 及时进行超声心动图筛查, 对于出现上述表现的PH患儿, 应注意MMA这一代谢病病因的检查, 以利早期诊断和早期治疗, 改善预后。
The authors have declared that no competing interests exist.
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|