
{kind=link}
{kind=link}
Williams-Beuren综合征的临床及遗传学特点:2例报道
[王姝琪1, 2 , 杨志仙1 , 李慧1  ]
]
]
|
|


通过分析2例WBS患儿临床表现,个人史,心脏超声、头颅磁共振成像、脑电图及染色体检测结果的特点,探讨Williams-Beuren综合征(Williams-Beuren syndrome,WBS)的临床及遗传学特点,以提高对该疾病的认识。2例患儿均为男性,就诊年龄分别为11个月1天和9个月9天,均存在心血管畸形,病例1为主动脉瓣上狭窄,病例2为房间隔缺损、动脉导管未闭。患儿具有WBS特征性面容:眶周饱满、球形鼻头、鼻梁低平、长人中、厚嘴唇。精神发育迟滞:病例1为中-重度,病例2为重度。其他表现:病例1合并双侧腹股沟斜疝和鞘膜积液,病例2合并喂养困难、埋藏式阴茎及婴儿痉挛。个人史:病例1孕期曾保胎,后因羊水污染足月剖宫产娩出。辅助检查:2例患儿头颅磁共振成像均未见明显异常,病例2脑电图示高度失律并监测到痉挛发作。染色体检测结果:病例1通过多重连接依赖的探针扩增法确定为染色体7q11.23缺失,其内包含人弹性蛋白(elastin, ELN)基因片段缺失突变;病例2通过高分辨G带法确定为染色体7q11.21q11.23缺失。本组2例WBS患儿均存在心血管畸形、特殊面容、精神发育迟滞及结缔组织或泌尿系统异常,病例1的主动脉瓣上狭窄可能与 ELN基因缺失相关,病例2的癫痫发生可能与超出7q11.23区域的q11.21缺失相关。
SUMMARY To explore the clinical and genetic characteristics of Williams-Beuren syndrome (WBS) and to raise awareness of the disease. The characteristics of clinical manifestations, personal history, cardiac ultrasound, brain magnetic resonance imaging (MRI), electroencephalogram (EEG) and chromosome detection results of two cases with WBS were analyzed. The two patients were both male and the age was 11 months and 1 day, and 9 months and 9 days, respectively. They both suffered from cardiovascular malformation: case one presented supravalvular aortic stenosis, and case two showed atrial septal defect and patent ductus arteriosus. Both of the cases were exhibited characteristic facial features of WBS, including full orbital, spherical nose, flat nasal bridge, long philtrum and thick lips. For the mental deve-lopment, case one displayed moderate to severe developmental retardation, and case two showed severe developmental retardation. In addition, case one presented bilateral indirect inguinal hernia and hydrocele, and case two manifested feeding difficulties, buried penis and infantile spasms. Personal history: case one’s mother had tocolytic therapy during pregnancy period, and case one was born at full-term by cesarean section due to amniotic fluid pollution. Supplementary examination: brain MRI of the two cases were no significant abnormalities, the EEG of case two showed hypsarrhythmia, and the epileptic spasms were recorded. Chromosome detection results: case one was identified as 7q11.23 deletion including the fragment deletion mutation of elastin ( ELN) gene by multiplex ligation dependent probe amplification method, and case two was found with 7q11.21q11.23 deletion by high resolution G-band method. The two cases with WBS both had cardiovascular malformations, special facial features, mental retardation and connective tissue or urinary system abnormality. The supravalvular aortic stenosis of case one may be associated with the deletion of ELN gene, and the occurrence of epilepsy of case two may be related to the q11.21 deletion beyond the 7q11.23 region.
Williams-Beuren综合征(Williams-Beuren syndrome, WBS)最早由Williams等[1]于1961年报道, 1962年Beuren等[2]也对这一疾病进行了报道, 因此得名为WBS。WBS在活产婴儿中的发病率约为1/7 500~1/20 000[3, 4], 是由7号染色体长臂近端(7q11.23)缺失导致, 常见缺失区域为1.55~1.84 Mb, 主要涉及包括人弹性蛋白(elastin, ELN)基因在内的20多个基因, 属常染色体显性遗传病, 但多数患儿为散发, 部分有家族史。
本病临床特征多样, 大多数患儿具有典型的WBS表现, 包括心血管系统疾病、特殊面容、生长发育迟滞、婴儿期一过性高钙血症和神经行为异常, 少数也可以是正常人[5]。本病目前尚无有效的治疗方法。近年来世界各地均有本病报道, 但国内此病报道较少。本文对2例确诊WBS的患儿进行了临床及遗传学特点分析, 以期进一步提高对本病的认识。
对2015年5月至2016年3月北京大学第一医院儿科神经门诊确诊的2例WBS患儿进行详细的病史采集, 包括临床特点、诊疗过程、个人史及家族史, 并通过神经门诊及电话随访至2016年7月。本研究获得北京大学第一医院临床研究伦理委员会批准, 患儿家长签署知情同意书。
2例患儿均进行血常规、血液生化、尿代谢筛查、头颅磁共振成像(magnetic resonance imaging, MRI)、脑电图及心脏超声检查。1例患儿进行了发育评估及血代谢筛查, 另1例患儿进行了血钙、尿钙及心电图检查。
病例1采用多重连接依赖的探针扩增法(multiplex ligation dependent probe amplification, MLPA)进行检测, 病例2采用染色体高分辨G带法进行检测。
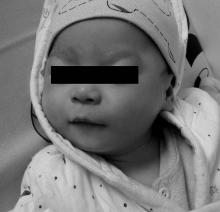
病例1, 男, 11个月1天, 因“ 发现精神运动发育落后7个月” 就诊。个人史:其母G3P1(表示孕3产1, 下同), G1人工流产, G2孕30天时自然流产, G3为本患儿, 因母孕1个月时孕酮低、阴道出血, 一直保胎至足月, 后因羊水混浊行剖宫产娩出, 患儿出生时无窒息, 出生体重2 950 g。家族史:家族中无类似病史。体格检查:身长68 cm[小于第3百分位(< P3)], 体重6 kg(< P3), 头围42.5 cm(< P3), 眶周饱满、球形鼻头、鼻梁低平、长人中、厚嘴唇(图1), 双侧腹股沟斜疝及鞘膜积液。精神运动发育:3个月竖头, 就诊时仍独坐不稳。
病例2, 男, 9个月9天, 因“ 反复抽搐4个月” 就诊。个人史:其母G3P2, G1人工流产, G2为现3岁的正常女童, G3为本患儿, 母孕期顺利, 足月顺产, 患儿出生体重3 050 g, 无宫内发育迟缓及宫内窘迫, 无羊水混浊, 出生时无窒息。新生儿期因“ 新生儿肺炎、病理性黄疸” 住院治疗, 后治愈出院。生后喂养困难, 4个半月左右出现痉挛发作, 最多时10串/天, 每串60~70下, 脑电图示高度失律并监测到痉挛发作, 诊断为婴儿痉挛。家族史:家族中无类似病史。体格检查:身长65 cm(< P3), 体重5.5 kg(< P3), 头围41.3 cm(< P3), 眶周饱满、球形鼻头、鼻梁低平、长人中、厚嘴唇、耳位低(图2), 左上眼睑下垂, 高腭弓, 埋藏式阴茎。精神运动发育:就诊时不能追光追物, 竖头不稳。
 | 图1 病例1, 男, 11个月1天, 特征性面容(眶周饱满、球形鼻头、鼻梁低平、长人中、厚嘴唇)Figure 1 Case one, male, 11 months and 1 day, the characteristic facial features (full orbital, spherical nose, flat nasal bridge, long philtrum and thick lips) |
 | 图2 病例2, 男, 9个月9天, 特征性面容(眶周饱满、球形鼻头、鼻梁低平、长人中、厚嘴唇)Figure 2 Case two, male, 9 months and 9 days, the characteristic facial features (full orbital, spherical nose, flat nasal bridge, long philtrum and thick lips) |
2例患儿的血常规、血液生化、代谢筛查及头颅MRI均未见明显异常。心脏超声:病例1为主动脉瓣上狭窄, 病例2为房间隔缺损、动脉导管未闭。脑电图:病例1未见明显异常, 病例2为高度失律、痉挛发作。染色体检测:病例1为46, XY, 7q11.23del, 其内包含ELN基因片段缺失突变, 病例2为46, XY, 7q11.21q11.23del。病例1发育评估为中-重度发育迟滞(Gessell发育量表:适应性、大运动、精细运动及个人-社交均为重度发育迟滞, 语言为中度发育迟滞), 病例2未进行量表发育评估(根据医生对患儿的观察及家长的描述粗略评估为重度发育迟滞)。病例2的血钙及尿钙检查正常, 心电图示S-T段抬高, 病例1未进行此项检测。
病例1于2015年7月(13月龄)行双侧腹股沟斜疝手术, 后失访; 病例2诊断为婴儿痉挛后曾给予多种药物治疗, 包括促肾上腺皮质激素(adrenocorticotrophic hormone, ACTH)肌肉注射28 d, 口服左乙拉西坦和托吡酯治疗, 痉挛发作无明显减少, 后行生酮饮食治疗2~3个月, 发作部分缓解(发作减少> 50%), 但因生酮饮食后患儿体重不增而停用。目前患儿已停用所有抗癫痫治疗(家长自行停用), 仍有痉挛发作, 2~4串/天, 每串20~30下, 偶有2~3天无发作。
WBS属常染色体显性遗传病, 1993年Ewart等[6]研究证实有主动脉瓣上狭窄(supravalvular aortic stenosis, SVAS)表现的WBS患儿是由7q11.23缺失导致。目前的研究表明, SVAS的发生是由于染色体异位缺失或点突变导致ELN基因的一个拷贝中断所致, 但其他与WBS临床表现相关的基因尚未确定[7, 8, 9]。
WBS是一个多系统紊乱的疾病, 临床表现多样且不特异, 常见的临床表现包括以下几个方面。
(1)以动脉狭窄为特征的心血管表现:约有80%的WBS患儿出现心血管畸形, 其中SVAS的发生率最高(占64%), 其次是肺动脉狭窄、主动脉缩窄、二尖瓣脱垂和动脉导管未闭等[10]。部分患儿早期的心脏检查可无明显异常, 但这不能证明心脏为正常状态, 特别是存在功能性杂音者, 由于心脏的病变已经存在, 随着年龄的增长很有可能进展为SVAS, 且SVAS也可随年龄的增长进行性加重, 导致左心室负荷逐渐加大、左心室肥大、心力衰竭乃至猝死, 有研究认为WBS患儿应定期进行心脏检查[11]。本组2例患儿均有心血管畸形, 其中病例1为SVAS, 符合典型WBS的表现; 病例2为房间隔缺损和动脉导管未闭, 虽未发现SVAS, 但因患儿年龄小, 随年龄增长有发展为SVAS的可能, 因此两例患儿均应定期随访心脏超声检查, 以观察疾病进展情况, 必要时及早干预。
(2)典型的面部特征:包括双颞狭小, 眶周饱满, 球形鼻尖、短而上翘的鼻子, 星状虹膜, 长人中, 厚嘴唇, 牙齿畸形或咬合不正。本组2例均有较为特征的面部表现(眶周饱满、球形鼻尖、长人中、厚嘴唇)。个别患儿也可无典型面部特征, 吕俊健等[12]曾报道1例以反复心律失常为主要表现且存在SVAS但不伴典型面容的WBS新生儿。对于不具有典型的特征性面容且没有SVAS的患儿, 早期可能会存在漏诊和误诊。
(3)运动发育迟缓或精神发育迟滞:文献报道WBS患儿常常存在不同程度的智力落后, 平均智商为50~60[13]。本组患儿均有发育迟滞, 病例1为中-重度发育迟滞, 病例2为重度发育迟滞。文献报道早期给予合适的刺激, 可提高患儿的智力水平[14], 因此对于本组患儿医生均建议其尽早开始康复训练。
(4)行为和认知的异常:包括过于友好, 同情他人, 焦虑, 对声音过敏, 在言语短时记忆和沟通能力的优势, 视觉空间结构的损伤等。本组两例患儿年龄尚小, 此方面的表现尚需随诊观察。
(5)内分泌系统问题:包括婴儿期一过性的高钙血症、糖尿病、亚临床甲状腺功能减退和性早熟等。一过性高钙血症的发生率约为30%[15], 病例2进行了血钙及尿钙检查均未见异常。
(6)结缔组织异常:包括声音嘶哑、腹股沟疝、脐疝等。本组的病例1有双侧腹股沟斜疝及鞘膜积液, 已行手术修复。
(7)肾功能异常和尿路缺损:包括肾功能不全、肾盂积水、肾结石、膀胱憩室、排尿障碍、尿路感染、隐睾和尿道下裂等。本组两例患儿均未见有肾脏的畸形, 但病例2合并有埋藏式阴茎。
(8)消化系统:可有婴儿期吸吮力弱、吞咽不协调、易呛、易呕吐、喂养困难、易便秘等表现。本组病例2存在喂养困难。
WBS的发生机制为染色体7q11.23区域内或染色体间低拷贝重复序列(low copy repeats, LCR)的重排。大约95%的WBS患儿存在7q11.23染色体区域的1.5~1.8 Mb缺失, 即典型缺失, 大约5%的患儿存在超出7q11.23区域的缺失(端粒外延部分), 即不典型缺失[16], 这些缺失中2/3是因为减数分裂过程中7号染色体间同源染色体重排所致, 1/3是由于染色体内的重排所致, 即此类患儿父母亲的WBS区域发生了臂内倒位[17]。本组中两例患儿的父母均未进行染色体相关检查, 通过孕产史资料分析, 病例1的母亲曾有1次胚胎早期的自然流产史, 且病例1在孕早期曾出现先兆流产, 推测该患儿的父母不排除有携带这种臂内倒位的可能。病例2的同胞姐姐正常, 其母无不良孕产史, 孕期也无明显异常, 故推测携带臂内倒位的可能性相对减小。
WBS缺失基因中与其临床表现关系最密切的是ELN基因, 文献报道96%的患儿存在ELN基因缺失[15], 它的表达产物是弹性蛋白, 是各种器官结缔组织中弹性纤维的重要成分。1993年, Ewart等[6]通过分析证实有SVAS表现的WBS患儿是由7q11.23缺失导致。Karmiloff-Smith等[13]研究认为ELN基因缺失与WBS的心血管病变(如SVAS)和结缔组织病变(如疝气)等相关, 而与其他临床表现无关。目前的研究表明, SVAS的发生是由于染色体异位缺失或点突变导致ELN基因的一个拷贝中断所致[7, 8, 9], 然而Li等[5]报道的一组患儿中, 有1例存在ELN基因缺失但没有心血管畸形和结缔组织异常, 另1例ELN基因正常的患儿却存在心血管和结缔组织畸形, 这提示可能存在其他导致心血管畸形的基因或因素。本组病例1存在SVAS心血管畸形, 染色体检查结果示7q11.23del, 并存在ELN基因片段缺失突变, 符合典型WBS表现。病例2虽不存在SVAS心血管畸形, 但合并有房间隔缺损和动脉导管未闭, 由于病例2采用的是染色体高分辨G带法, 对检测是否存在ELN基因缺失可能有一定限局性。
WBS常见缺失区域的基因还包括LIMK1、RFC2、STX1A、FZD3、GTF2I、CYLN2和GTF2IRD1等[18]。目前已知LIMK1基因可能与WBS的独特认知特点有关, GTF2IRD1基因主要影响患儿的特征性面容, 且GTF2IRD1和LIMK1都对视觉-空间构建异常起着重要的作用, CLIP2、GTF2I的缺失与大脑发育及社会行为认知能力等相关[19, 20]。本组患儿颅面部特征及智力障碍表现均很典型, 但由于年龄尚小, 是否会出现WBS独特的认知表现尚需随访观察, 必要时可进一步行相关基因检测以明确。
此外, 亦有国外文献报道, 不典型WBS患儿带有较大区域缺失, 且缺失的基因可能不同, 他们通常伴有严重的认知迟滞和癫痫, 包括婴儿痉挛[17, 21], 因此, WBS的神经系统表现也被认为与7q11.23区域的远端缺失有关(即7q11.23远端缺失综合征)[22]。国内既往尚未见WBS合并癫痫的病例报道, 国外已报道30余例, 其中9例为婴儿痉挛[16]。Mizugishi等[23]报道, 不典型7q11.23缺失包含的基因是WBS患儿发生婴儿痉挛的基础。Marshall等[24]的研究提示, MAGI2基因可能与婴儿痉挛有关。Ramocki等[22]报道了14例伴有癫痫和智力落后的WBS患儿, 其远端缺失片段中均包括HIP1基因。Fusco等[19]也发现有部分不典型缺失的患儿具有更明显的智力落后、孤独症和包括婴儿痉挛在内的癫痫, 所缺失的基因中包括HIP1和YWHAG基因。然而, Nicita等[16]的研究选取了7个癫痫中心中合并有癫痫的8例典型WBS患儿及1例7q11.23远端缺失综合征患儿, 结果显示该组患儿均未发现HIP1、YWHAG和MAGI2基因缺失。本组中病例2的染色体分析证实为不典型缺失, 临床合并有婴儿痉挛。基于以上研究提示, 病例2患儿的癫痫发生可能与超出7q11.23区域的q11.21缺失相关。
本组两例患儿的临床表现均存在心血管畸形、特殊面容、精神运动发育迟滞及结缔组织或泌尿系统异常, 提示为染色体异常相关疾病, 经进一步完善染色体检测证实为WBS。目前尚无法通过基因型来预测WBS患儿发生癫痫的可能性, 未来可进一步扩大样本量行基因检测, 以分析不典型缺失病例中婴儿痉挛发生的可能相关基因。
The authors have declared that no competing interests exist.
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|