
{kind=link}
{kind=link}
{kind=link}
{kind=link}
皮肌炎继发干燥综合征伴肺间质病变的血清人Ⅱ型肺泡细胞表面抗原变化1例
[余建峰1, 2, * , 金月波1, * , 何菁1  , 安媛
, 安媛1 , 栗占国1 ]
, 安媛]
|
|


SUMMARY Interstitial lung diseases (ILDs) are a diverse group of pulmonary disorders characterized by various patterns of inflammation and fibrosis in the interstitium of the lung. The underlying pathogenesis of ILDs is complex and associated with multiple rheumatologic conditions, such as systemic sclerosis, rheumatoid arthritis, pollymyositis and dermatomyositis, Sjögren’s syndrome, and systemic lupus erythematosus. As the disease progresses, excessive pulmonary fibrosis impairs alveolar gas exchange and damages pulmonary function. The common methods to diagnose ILDs, such as clinical manifestations, pulmonary function test, and radiological examinations are not specific for ILDs and not able to diagnose ILDs at the early stage due to their low sensitivity. So, the easy way is important to diagnose ILDs. One important biomarker for ILDs is the high-molecular-weight glycoprotein, Krebs von den Lungen-6(KL-6). KL-6 encoded by the MUC1 gene is a mucin-like glycoprotein with high molecular weight and expressed predominantly on the cell surface of type Ⅱ alveolar epithelial cells, and is detectable in the serum of patients with ILDs. We here report a case of ILDs associated with dermatomyositis and secondary Sjögren’s syndrome. A 60-year-old woman was admitted to our hospital with the chief complaints of debilitation, dry mouth, dyspnea and astasia. ILDs associated with dermatomyositis and secondary Sjögren’s syndrome was diagnosed clinically when the following criteria were satisfied: (1) development of dyspnea within 2 months of presentation, (2) pulmonary dispersion dysfunction, (3) bilateral infiltrative shadows on chest high resolution computed tomography (HRCT). She was treated with prednisone 50 mg/d prior to admission, but the result of therapy was not good. In our hospital she was treated with intravenous methylprednisolone and cyclophosphamide and oral hydroxychloroquine sulfate. Subsequently, her serum KL-6 levels gradually decreased after treatment, pulmonary diffuse function improved, and the improvement in the clinical manifestation and HRCT findings were observed. Nevertheless, the combination treatment of glucocorticoid and cyclophosphamide had contributed to the favourable outcomes. In conclusion, detection of serum KL-6 levels in ILDs associated with connective tissue diseases may be be-neficial to making a definitive diagnosis, predicting the prognosis and monitoring the disease activity, which would be of great help in clinical practice. However, a well-designed clinical study with more patients and a longer follow-up period are required to arrive at a more conclusive judgment on the role of serum KL-6 in patients with ILDs.
自身免疫性疾病往往可继发多脏器多系统损害, 肺是常见受累器官之一, 而自身免疫性疾病导致的肺间质病变(interstitial lung diseases, ILDs), 往往以肺间质和肺泡腔不同形式和程度的炎症及纤维化为主要病理改变, 病情进展迅速, 最终结局为呼吸衰竭。常见的与ILDs相关的自身免疫性疾病有系统性硬化症、类风湿关节炎、系统性红斑狼疮、原发性干燥综合征、多发性肌炎/皮肌炎等。文献报道, 一种高分子量糖蛋白— — 人Ⅱ 型肺泡细胞表面抗原(Krebs von den Lungen-6, KL-6)与上述疾病所致的ILDs关系密切。KL-6在正常肺组织的Ⅱ 型肺泡细胞表达, 当发生ILDs时, Ⅱ 型肺泡细胞再生, KL-6明显升高, 不仅出现于ILDs患者的肺泡灌洗液中[1], 同时也存在于血清中[2]。现报道北京大学人民医院风湿免疫科的1例自身免疫性疾病合并ILDs的患者治疗前后血清KL-6的变化情况, 并结合临床和影像学特点来评估病情, 指导治疗。
患者女性, 60岁, 主因“ 乏力、口干、活动后气短、起立困难2月余” 入院。入院前2月余以双下肢乏力、口干起病, 逐渐出现活动后气短、心悸、消瘦及小腿肌肉酸胀不适, 下蹲后起立困难, 外院查抗核抗体(antinuclear antibody, ANA)、抗U1核糖核蛋白抗体(anti-U1 ribonuclear protien antibody, 抗U1RNP抗体)、类风湿因子(rheumatoid factor, RF)阳性, 肌酸激酶(creatine kinase, CK)、谷丙转氨酶(alanine aminotransferase, ALT)、谷草转氨酶(aspartate transaminase, AST)、γ -谷氨酰转移酶(γ -glutamyl transpeptidase, γ -GT)升高, 肺部X线计算机断层摄影(computed tomography, CT)示ILD, 当地考虑“ 结缔组织病合并ILD” , 予醋酸泼尼松50 mg, 每日1次, 治疗2周, 疗效欠佳, 入北京大学人民医院风湿免疫科进一步诊治。既往史、个人史、家族史等无特殊。
入院查体:舌体发红, 舌乳头消失, 双下肺可闻及Velcro啰音。全身皮肤未见皮疹, 全身关节无肿胀、畸形及压痛, 四肢肌力Ⅳ 级, 肌张力正常, 全身肌肉无压痛。
入院后完善检查:血、尿、便常规均正常。生化指标:ALT 73 U/L, AST 52 U/L, CK 303 U/L, 血清白蛋白(serum albumin, Alb) 39.0 g/L, 血清尿酸(uric acid, UA) 368 μ mol/L。C-反应蛋白(c-reactive protein, CRP)、红细胞沉降率(erythrocyte sedimentation rate, ESR)、免疫球蛋白均正常。补体C4 0.131 g/L, RF 209.0 IU/ML。自身抗体:ANA滴度为1 :640斑点型, 抗核糖核蛋白抗体(anti-ribonuclear protien antibody, 抗RNP抗体)滴度为1 :10 000, 抗SSA52抗体(anti Sjö gren’ s syndrome A52 antibody)阳性, 抗 SSA60(anti Sjö gren’ s syndrome A60 antibody)抗体、抗SSB抗体(anti Sjö gren’ s syndrome B antibody)、抗SCL-70抗体(anti scleroderma-70 antibody)、抗Sm抗体(anti Smith antibody)、抗Rib抗体(anti ribosomal antibody)均阴性。肌炎谱:抗SRP抗体(anti-signal recognition particle antibody)(++)。动脉血气分析:血液酸碱度 7.441, 二氧化碳分压 38.4 mmHg(1 mmHg=0.133 kPa), 血氧分压 80.1 mmHg, 血氧饱和度 96.8%, 氧和指数383.4 mmHg。真菌G试验[(1, 3)-beta-D-glucan, G test]:220.3 ng/L。巨细胞病毒DNA、真菌GM试验(galacto-mannan, GM test)、乙型肝炎表面抗原、丙型肝炎抗体、获得性免疫缺陷综合征病毒抗体、梅毒螺旋体抗体以及肿瘤标记物均阴性。干眼症检查:角膜荧光素染色:右眼(-)、左眼(-); 泪膜破裂试验:右眼8 s、左眼9 s; 泪液分泌实验:右眼大于20 mm/5 min、左眼大于30 mm/5 min。血清KL-6 1 850 U/mL(本实验室正常人参考值为≤ 500 U/mL)。超声心动检查:左心室舒张功能减退, 左心室射血分数67.4%。腹部超声检查:脂肪肝。胸部CT(图1):双肺间质性肺炎。肺功能测定提示:一口气弥散量为4.52 mmol/(min· kPa), 一口气弥散量实际值/预测值为59.1%。支气管镜检查:气管支气管末未见异常。灌洗右中叶内侧段, 支气管灌洗液细胞计数及分类:细胞总数0.15× 109/L, 巨噬细胞74.00%, 淋巴细胞 24.00%, 分叶核细胞2.00%, 嗜酸性粒细胞、嗜碱性粒细胞均未见。支气管灌洗液病原学检查均未见异常。(右中叶)支气管镜灌洗液涂片镜检(图2):可见大量退变的肺泡上皮细胞、吞噬细胞及淋巴细胞, 少量中性粒细胞, 散在红细胞, 个别鳞状上皮细胞。肌电图:(1)神经电图检查:神经源性损害(右侧正中神经为主); (2)针极电图检查:肌源性损害(右侧三角肌)。肌肉病理(左肱二头肌)诊断:骨骼肌的主要病理改变是散在再生肌纤维, 肌纤维膜主要组织兼容性复合物阳性表达及肌纤维补体沉积, 符合坏死性肌病的病理改变, 可出现在免疫性坏死性肌肉病中。唇腺活检病理(右下唇, 图3):小涎腺组织, 小叶及腺泡结构存在, 腺泡结构未见明确破坏, 个别导管扩张, 周围可见灶状淋巴细胞, 每光镜视野2 mm× 2 mm下, 浆细胞浸润> 50个, IgG(局部+), IgG4(-)。
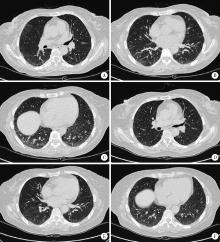
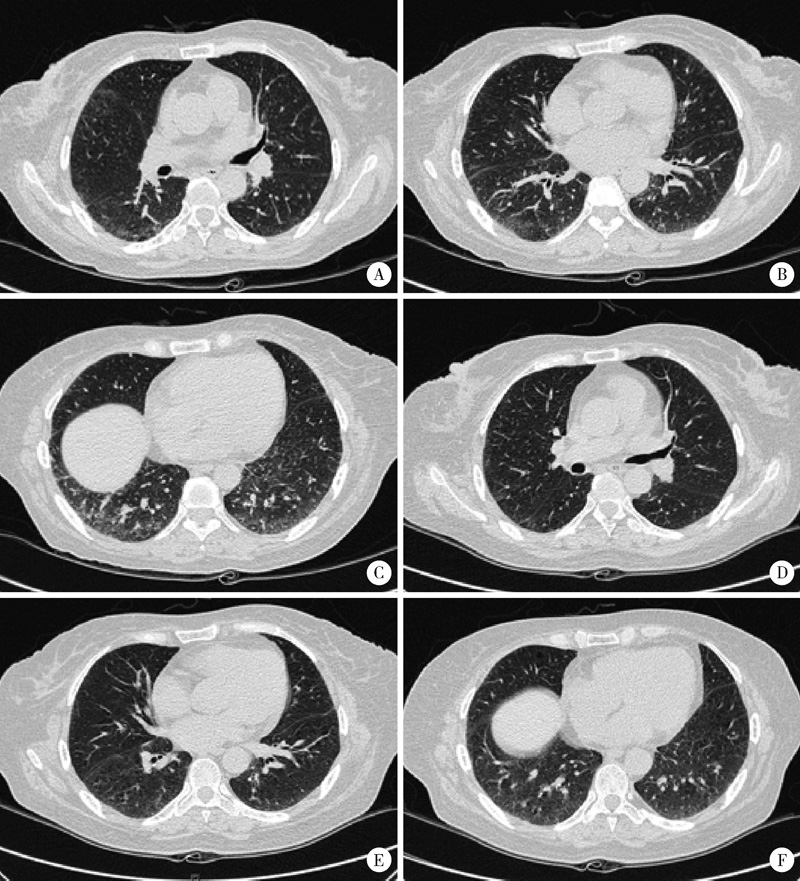 | 图1 患者肺部CT图像Figure 1 CT scan of the chest A-C, diffuse scattered spots, ground-glass shadow and a little consolidation of lung tissue were located in the subpleural area of both lungs before treatment(Jun. 15, 2015); D-F, right lung lesions were improved after treatment(Aug. 7, 2015). |
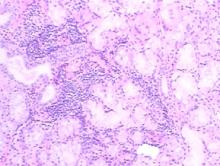
 | 图2 支气管肺泡灌洗, 可见大量退变的肺泡上皮细胞、吞噬细胞及淋巴细胞, 少量中性粒细胞, 散在红细胞, 个别鳞状上皮细胞(HE × 400)Figure 2 Bronchoalveolar lavage, histopathological findings showed a large number of degenerative alveolar epithelial cells, phagocytes, lymphocytes, and a few neutrophils, dispersed erythrocytes, and rare squamous epithelial cells(HE × 400) |
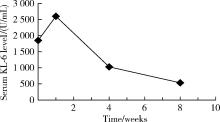
入院予甲泼尼龙200 mg 每日1次, 强化治疗3 d, 环磷酰胺400 mg冲击1次, 以及羟氯喹0.2 g, 每日2次口服治疗, 同时辅以保肝、补钙等对症治疗。治疗1周后复查肺部CT提示右肺间质性改变较前加重, 血清KL-6结果为2 602 U/mL, 继续给予甲泼尼龙500 mg , 每日1次, 冲击治疗5 d, 并予环磷酰胺400 mg, 每2周1次, 连续冲击2次。1个月后患者诉症状较前明显好转, 复查胸部CT(图1)提示ILD较前减轻, 血气分析提示血氧分压 87 mmHg, 肺功能测定提示:一口气弥散量为5.58 mmol/(min· kPa), 一口气弥散量实际值/预测值为73.3%。复测血清KL-6为1 030 U/mL, 患者病情好转出院。出院诊断:免疫介导的坏死性肌病, 继发性干燥综合征, 肺间质病变。出院后继续环磷酰胺 400 mg, 每2周1次冲击, 联合醋酸泼尼松50 mg 每日1次治疗。再过1个月后返院, 患者查体可见右手近端指间关节及掌指关节伸面、左手近端指间关节伸面以及双侧肘关节伸面皮肤红色斑疹, 胸前弥漫性暗红色斑疹, 考虑患者新发皮疹特点符合皮肌炎皮疹表现, 遂修正诊断为:皮肌炎, 继发性干燥综合征, 肺间质病变。再次复查肺部CT提示部分病变较前进一步减轻, 血气分析示血氧分压93.7 mmHg, 复测血清KL-6降至535 U/mL(图4)。然后患者出院并继续醋酸泼尼松规律治疗, 并按时减量, 同时联合环磷酰胺400 mg, 每2周1次冲击治疗。出院后随访1个月病情平稳。
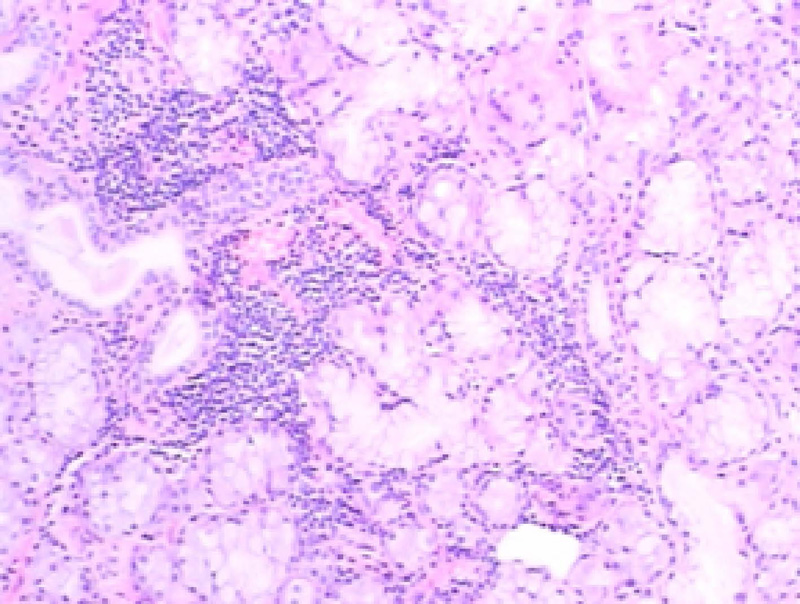 | 图3 唇腺活检病理, 可见小涎腺组织, 小叶及腺泡结构存在, 腺泡结构未见明确破坏, 个别导管扩张, 周围可见灶状淋巴细胞、每光镜视野2 mm× 2 mm下浆细胞浸润> 50个, IgG (局部+), IgG4(-)(HE × 400)Figure 3 Labial salivary gland histopathological findings showed minor salivary glands, lobules and acinus structures without destruction and rare dilation of salivary gland ducts, which were infiltrated by focal lymphocytes and plasma cells> 50 cells per 2 mm× 2 mm under light microscope, partial IgG(+), IgG4(-)(HE × 400) |
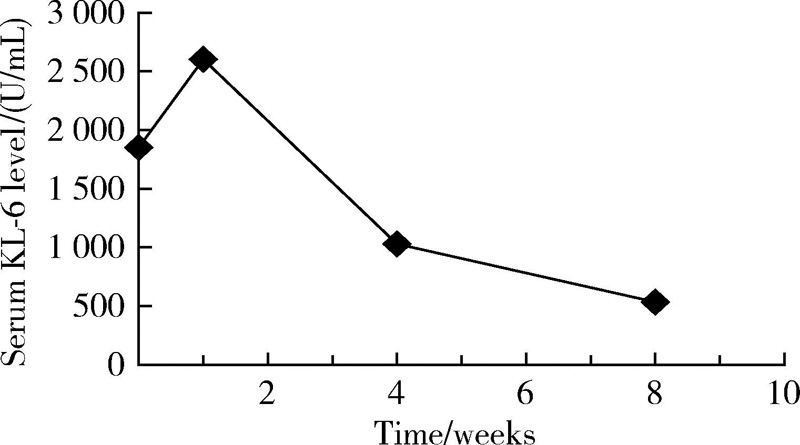 | 图4 治疗前、治疗后1周、治疗后4周及治疗后8周血清KL-6的变化Figure 4 Changes of serum KL-6 levels before the treatment, 1 week, 4 weeks and 8 weeks after treatment |
临床上诊断ILDs后, 在治疗过程中往往需要通过观察症状、体征, 并通过高分辨CT、支气管肺泡灌洗、肺功能等检查来判断疾病活动度, 预测疾病预后, 然而症状和体征往往没有特异性, CT对人体有一定的电离辐射, 支气管镜检查则不同程度地引起患者的不适, 肺功能无法全面反映肺结构损伤。
尽管血清标记物如CRP、ESR、乳酸脱氢酶、表面蛋白-A(surfactant protein A, SP-A)、表面蛋白-D(surfactant protein D, SP-D)等可以一定程度上反映疾病情况, 但不具有特异性, 上述指标的升高并不能区分细菌性肺炎和间质性肺炎, 临床上亟待开发一种便捷、廉价、伤害小而特异性高的方法来取代上述检查。KL-6是30年前日本学者[3]研究肺腺癌时发现的一种大分子糖蛋白抗原物质, 最初是作为一种肿瘤标志抗原进行研究。KL-6可表达于肺的Ⅱ 型肺泡上皮细胞、支气管腺上皮细胞、以及正常细胞如胰腺、乳腺导管中, 在正常肺组织和终末肺支气管肺泡上皮细胞上只有极少量的表达, 在肺泡Ⅰ 型上皮细胞上几乎不表达。在肺泡损伤的初期, 肺泡基底膜Ⅱ 型上皮细胞再生, KL-6表达明显增加, 可被释放于肺泡灌洗液及血清中[4]。尽管血清KL-6不能直接反映炎症损害的程度, 但可提示肺泡上皮细胞和肺间质渗透力的损伤程度。在轻度肺损伤的患者中, 可以观察到SP-A、SP-D短期内升高, 但KL-6无明显变化[5]。近年来越来越多的研究表明, 血清KL-6与ILDs有较大相关性。2012年Fathi等[6]学者发现在白种人肺间质病变的患者中, 其血清KL-6值明显高于正常人。随后又有中国学者[7]分别检测了373例ILDs患者、584例非ILDs的肺病患者以及127例健康志愿者的血清KL-6, 发现ILDs患者的KL-6水平显著高于后两者水平, 提示KL-6可对ILDs的诊断提供帮助。进一步的分析则发现血清KL-6可反映出疾病的严重程度。同时, 此研究[7]观察了其中106例ILDs患者治疗前后血清KL-6变化, 发现其中58.5%的患者治疗效果较好, 且治疗后血清KL-6较前明显降低, 提示KL-6可随着疾病好转而降低, 随着疾病恶化而升高。同一结果也可见于Ohshimo等[8]所做的研究, KL-6随着疾病的进展而显著升高, KL-6可能为ILDs是否进展的预测因子。Honda等[9]发现, 在结节病患者中, 较重的CT改变往往伴随着更高的血清KL-6值。Kinoshita等[10]也认为, 在类风湿关节炎合并ILDs的患者中, CT的病变程度与血清KL-6呈正相关。有学者[11]观察了100例多肌炎/皮肌炎患者, 将其分为两组, 一组合并ILDs, 共计56例, 另一组无肺部受累, 共计44例, 进一步监测所有患者血清KL-6、单核细胞趋化因子蛋白-1、SP-A及SP-D的浓度, 两组比较发现, 尽管合并ILD组患者的血清KL-6、SP-A、SP-D明显高于无ILD组, 但只有KL-6会随疾病进展而升高, 具有预测价值, 其余3种则与疾病进展无明显关系, 同时发现, KL-6水平可随患者用力肺活量及肺一氧化碳弥散量的好转而降低。
对于KL-6参考值的设定, 早在2006年就有学者[12]发现血清KL-6与MUC1基因(mucin 1 gene)多态性高度相关, 白种人高表达KL-6的基因型所占比例高于日本人, 故不同人种应设定不同的KL-6参考值。中国学者[7]在研究中将正常参考范围设定为KL-6< 500 U/mL, 血清KL-6> 500 U/mL则为阳性结果。由此计算出KL-6诊断ILD的特异性及敏感性分别为94.04%、77.75%, 认为500 U/mL作为分界线较适合于中国人区分ILD与非ILD, 而日本学者[13]也认为将临界值设定为500 U/mL, 能较好区分ILD患者与非ILD或健康人。
本例患者特点:(1)亚急性病程, 起病时有双下肢肌肉酸痛不适, 舌体干燥、疼痛不适, 伴活动后喘息, 查体可闻及双下肺Velcro啰音, 四肢肌力肌张力基本正常。(2)辅助检查发现ANA、抗SSA52、抗RNP抗体、抗SRP抗体均阳性, RF和KL-6明显升高, 肺部CT示双肺间质性肺炎; 支气管灌洗液:巨噬细胞74.00%, 淋巴细胞24.00%, 分叶核细胞2.00%; 肌肉活检提示:坏死性肌病病理改变, 再生肌纤维, 肌纤维膜主要组织相容性复合体-1阳性表达及肌纤维补体沉积。唇腺活检:灶状淋巴细胞, 每光镜视野2 mm× 2 mm下浆细胞浸润> 50个。(3)最后诊断为:皮肌炎, 干燥综合征, 继发性肺间质病变, 治疗上行甲泼尼龙冲击治疗、环磷酰胺冲击治疗联合羟氯喹口服, 治疗后患者临床表现、一口气弥散量、CT表现较前明显好转, 且血清KL-6较前明显下降。血清KL-6变化与肺部CT改变呈正相关, 与文献[11]所述相符。
本例初发病时并非典型皮肌炎, 后期随着病情进展, 才逐渐出现皮肌炎典型皮疹, 故诊断皮肌炎, 且合并干燥综合征。本例在不能明确典型皮肌炎诊断的情况下, 及时监测血清KL-6并结合肺部CT结果, 对于判断病情, 确定治疗以及预测预后, 有一定的临床意义。综上所述, 血清KL-6在本例病例中体现如下价值:诊断疾病; 评估疾病活动度, 判断治疗效果; 预测疾病预后。相信未来随着临床研究样本的扩大, KL-6与ILD的关系会日趋明朗, 也会被更多国内的医生所认识, 可以协助诊断和评估疾病情况。
The authors have declared that no competing interests exist.
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|