





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
沉默液泡型ATP酶c亚基 ATP6V0C抑制人前列腺癌细胞侵袭的分子机制
[邹鹏程1, 2 , 杨一峰1 , 徐晓艳1, 3, 4 , 刘北英5 , 梅放1 , 由江峰1 , 刘启忱6 , 裴斐1, △  ]
]
]
|
|


目的 采用RNA干涉技术沉默液泡型ATP酶c亚基 ATP6V0C的表达,探索ATP6V0C在前列腺癌侵袭中的分子作用机制,并研究其与LASS2/TMSG1的关系。方法与结果: ATP6V0C在高转移潜能人前列腺癌细胞系(PC-3M-1E8和PC-3M)中的表达明显高于低转移潜能人前列腺癌细胞系(PC-3M-2B4和PC-3), 选择ATP6V0C表达量最高的PC-3M-1E8细胞进行基因沉默实验,发现ATP6V0C的siRNA转染PC-3M-1E8后,其基质金属蛋白酶2(matrix metalloprotein-2,MMP-2)和基质金属蛋白酶9(MMP-9)蛋白的表达量及MMP-2的分泌量均无明显变化,但上清液中MMP-9的活性明显减弱,与空白对照组及阴性对照组相比,差异有统计学意义( P<0.05);干扰组细胞外氢离子浓度及液泡型ATPase的活性明显降低( P<0.05);细胞划痕修复实验及transwell体外侵袭实验结果显示 ATP6V0C siRNA干扰组细胞的体外迁移及侵袭能力明显减弱( P<0.05)。PC-3M-1E8细胞转染 ATP6V0C siRNA后,LASS2/TMSG1的表达明显减弱( P<0.05)。免疫荧光双重染色结果显示ATP6V0C蛋白与LASS2/TMSG1共定位于胞浆和胞膜,干扰组共定位信号与对照组相比明显减弱。结论 特异性siRNA沉默 ATP6V0C能抑制人前列腺癌细胞的迁移和侵袭能力,其机制与 ATP6V0C沉默后抑制V-ATPase的活性,使细胞外氢离子浓度降低,抑制MMP-9的激活,从而影响细胞外基质的降解和重塑有关。靶向siRNA干扰ATP6V0C的表达后,可能通过反馈调节机制抑制LASS2/TMSG1的表达,但其具体分子机制尚待进一步研究。
Objective: Vacuolar ATPase (V-ATPase) was found within the membranes and internal organelles of a vast array of eukaryotic cells, and was related to various kinds of highly metastatic tumors. LASS2/TMSG1 gene was a novel tumor metastasis suppressor gene cloned from human prostate cancer cell line PC-3M in 1999 by our laboratory. It was found out that protein encoded by LASS2/TMSG1 could interact with the c subunit of V-ATPase ( ATP6V0C). In this study, To use RNA interference to suppress the expression of ATP6V0C and try to further investigate the molecular mechanism of ATP6V0C in tumor metastasis and its relationship with LASS2/TMSG1 gene.Methods andResults: The expression level of ATP6V0C mRNA and protein in high metastatic potential prostate cancer cell lines (PC-3M-1E8 and PC-3M) was significantly higher than that in low metastatic potential prostate cancer cell lines (PC-3M-2B4 and PC-3), the expression level in PC-3M-1E8 being the highest. Follow-up tests selected PC-3M-1E8 cells for gene silencing. The expression and secretion of MMP-2 and the expression of MMP-9 in ATP6V0C siRNA transfected PC-3M-1E8 cells displayed no obvious change, but the activity of secreted MMP-9 was abated noticeably compared with the controls ( P<0.05). Extracellular hydrogen ion concentration and V-ATPase activity in interference group were both reduced significantly compared with the controls ( P<0.05). The migration and invasion capacity of ATP6V0C siRNA interfered cells in vitro were diminished significantly compared with the controls ( P<0.05). Furthermore, a dramatic reduction of LASS2/TMSG1 mRNA and protein level after transfection of siRNA in PC-3M-1E8 cells was discovered ( P<0.05). Confocal immunofluorescence showed a vast co-localization of ATP6V0C protein and LASS2/TMSG1 protein in plasma and membrane. The co-localization signals of control group were much stronger than those of interference group.Conclusion: Specific siRNA silencing of ATP6V0C gene inhi-bits the invasion of human prostate cancer cells in vitro by mechanism of inhibiting V-ATPase activity and then reducing the extracellular hydrogen ion concentration, inhibiting MMP-9 activation and affecting ECM degradation and reconstruction. Meanwhile, ATP6V0C and LASS2/TMSG1 have interaction and it is likely that ATP6V0C functions as a feedback regulator of LASS2/TMSG1.
前列腺癌已经成为威胁老年男性健康的主要恶性肿瘤, 在欧美国家(尤其是美国)的发病率最高, 而在亚洲地区(尤其是我国)发病率较低。随着人均寿命的延长和环境污染的加重, 近年来我国前列腺癌的发病率有明显上升的趋势, 在男性泌尿生殖系统恶性肿瘤中发病率跃居第3位。临床回顾性研究表明[1], 我国20世纪90年代前列腺癌的发病率较50年代增长5倍。目前, 前列腺癌发生转移往往不可治愈, 是导致前列腺癌死亡的主要原因, 因此寻找前列腺癌转移的相关基因和治疗靶点, 有效抑制前列腺癌细胞的生长和增殖, 是前列腺癌防治领域研究的重点之一[2]。
真核细胞中广泛存在的液泡型ATP酶(vacuolar ATPase, V-ATPase)在多种高转移性肿瘤中高表达, 并与肿瘤的侵袭和转移密切相关[3]。V-ATPase是一种ATP依赖性的质子泵, 由两个结构域组成, 分别称为V1和V0, V1亚单位由8个亚基组成, 主要作用是水解ATP并释放能量; V0亚单位由5个亚基组成, 主要起到质子运输通道的作用[4]。V0亚单位的c亚基, 又称ATP6V0C, 是V-ATPase质子泵的关键跨膜部分, 可以逆浓度梯度将H+泵至细胞囊腔中或细胞外, 维持细胞内的相对中性环境和细胞囊泡腔与细胞外的相对酸性环境。目前对V-ATPase的研究主要集中在肝癌和乳腺癌领域, 而对前列腺癌的研究较少见。在前列腺癌转移相关基因中, LASS2/TMSG1基因(tumor metastasis suppressor gene-1)是北京大学基础医学院病理学系, 于1999年12月应用mRNA差异显示技术, 从人前列腺癌细胞系PC-3M的不同转移潜能亚系中首先克隆出的一种新的肿瘤转移抑制基因[5], 通过酵母双杂交和GST pull-down实验发现, 它可以与V-ATPase的 c亚基ATP6V0C直接结合[6, 7], 推测其抑制肿瘤转移的作用机制可能与LASS2/TMSG1蛋白结合ATP6V0C后抑制了V-ATP酶的活性有关[8], 而V-ATPase活性降低可以抑制肿瘤细胞的生长、侵袭和转移[9]。国内外研究主要是针对LASS2/TMSG1对V-ATPase活性的作用, ATP6V0C是否对LASS2/TMSG1具有反馈调节作用目前尚未见相关报道。
本研究检测了不同转移潜能前列腺癌细胞系中ATP6V0C mRNA和蛋白的表达, 并为后续的基因沉默筛选了合适的细胞系。同时, 为了进一步探讨ATP6V0C在前列腺癌侵袭中的分子作用机制, 本研究采用ATP6V0C siRNA转染人前列腺癌高转移潜能细胞系PC-3M-1E8(ATP6V0C高表达), 并检测干扰前后该细胞V-ATPase的活性及细胞外氢离子浓度的改变, 以及基质金属蛋白酶的表达、分泌及活性的变化, 同时观察RNA干扰对PC-3M-1E8细胞体外迁移和侵袭能力的影响, 从而为判断前列腺癌侵袭、转移及预后提供新的分子生物学标志物, 并可能成为药物治疗的新靶点。
Trizol Reagent和脂质体LipofectamineTM2000 Reagent购自美国Invitrogen公司, TransScript First-Strand cDNA Synthesis SuperMix购自美国TransStart公司; Transwell小室(内径 6.5 mm, 孔径8.0 μ m) 购自美国Corning 公司; MatrigelTM 购自美国 BD 公司; 羊抗人多克隆抗体ATP6V0C和LASS2 均购自美国Santa Cruz公司; 鼠抗人单克隆抗体金属蛋白酶2(matrix metalloprotein-2, MMP-2)购自美国CST公司; V-ATPases 活性检测试剂盒购自上海GEMEND公司; BCECF氢离子敏感荧光探针购自美国Molecular Probes 公司。
针对人ATP6V0C基因mRNA 序列(NM001694)设计及合成4种靶向ATP6V0C的特异性干扰片段siRNA, 便于验证沉默效果的特异性, 其中siRNA-1针对的靶序列是5'-CACAAAGTAGACCCTCTCCGA-3', 对应 NM001694的第693~713位的核苷酸; siRNA-2 针对的靶序列是5'-CCCACCAGCCACAG-AATATTA-3', 对应NM001694的第715~735位核苷酸; siRNA-3 针对的靶序列是 5'-TGCGCGGAGCTGTGTCCAATA-3', 对应 NM001694的第1139~1159位核苷酸; siRNA-4 针对的靶序列是5'-GCGGATGATTTAGAATTGTCA-3', 对应NM001694的第952~972位核苷酸。以上4种靶向LASS2的特异性干扰片段siRNA及无关阴性对照组siRNA(Unc)均由美国 Qiagen 公司设计及合成, 该公司提供的siRNA是通过 BioPredsi运算后进一步经过一系列严格的分析获得的, 基因沉默结果的重复性好。
高转移潜能前列腺癌细胞系(PC-3M-1E8和PC-3M)和低转移潜能前列腺癌细胞系(PC-3M-2B4和PC-3)由北京大学基础医学院病理学系肿瘤生物学研究室提供, 用含10% (体积分数)小牛血清的1640培养液, 于37 ℃ 5% (体积分数) CO2孵育箱中培养。
取对数生长期细胞接种于6孔细胞培养板中, 3× 105个细胞/孔, 常规培养24 h。将5 μ L LipofectamineTM2000分别与2 μ mol/L的siRNA-1、siRNA-2、siRNA-3、siRNA-4 及 Unc 50 μ L配成转染混合物加入细胞中, 同时用PBS处理的细胞作为空白对照, 于37 ℃ 5%(体积分数) CO2的条件下培养。转染4~6 h后, 弃培养液, 加入含10% (体积分数)小牛血清的 RPMI1640培养液继续培养24 h。
细胞转染24 h后, 按照Trizol试剂盒进行抽提细胞总RNA, 并按TransScript First-Strand cDNA Synthesis SuperMix for RT-PCR试剂盒说明书对其进行反转录合成cDNA。按照 TransStart Green qPCR SuperMix试剂盒说明书进行PCR反应, GADPH、LASS2/TMSG1和ATP6V0C的引物经GenBank检索, 利用Primer 5.0软件设计, 由北京天一辉远生物公司合成。引物序列如下:GAPDH-A为5'-GGTCACCAGGGCTGCTTTTA-3', GAPDH-AS为5'-TCCTGGAAGATGGTGATGGG-3'; ATP6V0C-A为5'-ATGTAAAGACCACCCCTCCT-3', ATP6V0C-AS为5'-GA-AACAGACGATGGGCACTA-3'; LASS2/TMSG1-A为5'-CCTCTGATGTCAAGCGAAAG-3', LASS2/TMSG1-AS为5'-GCAGGTAATCGGAAGAGTCA-3'。PCR反应条件: 95 ℃预变性5 min; 95 ℃ 15 s, 60 ℃ 15 s, 72 ℃ 30 s, 共45个循环; 溶解曲线反应条件为 95 ℃ 15 s, 60 ℃ 30 s, 95 ℃ 15 s。实验重复3次。
实验细胞转染24 h后, 消化并转接种于35 mm培养皿中, 3× 105个细胞/皿, 细胞贴壁后换无血清1640培养液500 μ L继续培养24 h, 收集上清液, 同时提取细胞总蛋白。采用 BCA 法对蛋白进行总定量, 检测蛋白浓度, 调整平衡蛋白量。样品与2× SDS上样缓冲液等体积混合, 沸水浴 5 min, 取10 μ g上样。以β -actin为内参, 行10% (体积分数) SDS-PAGE电泳、转膜、封闭后, 一抗[β -actin工作浓度1 ∶ 2 500(体积比)稀释, ATP6V0C工作浓度1 ∶ 100(体积比)稀释, LASS2/TMSG1工作浓度 1 ∶ 2 000(体积比)稀释, MMP-2工作浓度1 ∶ 1 000(体积比)稀释, MMP-9工作浓度1 ∶ 500(体积比)稀释], 4 ℃培育过夜; PBST洗膜, 二抗[工作浓度 1 ∶ 5 000(体积比)稀释] 室温下培育1 h; PBST(phosphate buffer solution with Tween-20)洗膜, 按ECL化学发光试剂盒的说明书发光、压片。
用上海杰美公司提供的V-ATP酶活性检测试剂盒来检测细胞内V-ATP酶的活性, GEMEND动物线粒体呼吸链复合物(V-ATP酶)活性定量检测试剂是一种旨在使用丙酮酸激酶和乳酸脱氢酶连续循环法的反应系统, 测定与ATP合成对应的ATP水解产生ADP过程中伴随的 NADH(nicotinamide adenine dinucleotide)的氧化反应, 即采用比色法使用紫外分光光度仪测定其氧化后峰值(NADH浓度)的变化, 以此测算单位酶活性。3组实验细胞的V-ATP酶活性按照该试剂盒的检测步骤进行检测, 实验重复3次。
将指数生长期的细胞接种于六孔板中, 12 h后细胞密度达到30%~50%。分别转染特异的ATP6V0C的干扰片段及阴性对照干扰片段, 转染48 h后更换细胞培养液, 每孔中分别加入500 μ L 无血清、无 H +/HC缓冲体系、pH为7.0的1640培养液继续培养, 在第8、12和24小时各吸取上清液100 μ L, 然后于上清液中加入终浓度为1 μ mol /L的BCECF氢离子敏感荧光探针, 用荧光分光光度计分别检测在激发光波长为490 nm和440 nm处、发射光波长为535 nm处的光密度(D)值, 计算490 nm/440 nm激发下535 nm的发射光比值, 此比值(FIR)与 BCECF 浓度无关而仅与pH相关[10]。实验证明, 在pH 5~8范围内, FIR与pH呈线性关系, 将得到的细胞外pH值转换成细胞外氢离子浓度。实验重复3次。
六孔板转染细胞48 h后, 倒掉有血清培养液, 每孔加入500 μ L无血清1640培养液继续培养, 24 h后取细胞上清液, 4 ℃ 12 000 r/min 离心5 min, 并用BCA法测定上清液中的蛋白浓度。将上清液与5× 上样缓冲液[不能含有DTT(DL-dithiothreitol)]混合, 煮沸10 min, 然后行10%(质量分数) SDS-PAGE (含质量分数0.1% A型明胶)电泳, 每泳道蛋白上样量为20 μ g, 120 V恒压电泳2 h。电泳后将凝胶置于洗脱液(质量分数2.5 % Triton X-100, 50 mmol/L Tris-Cl pH=7.5, 5 mmol/L CaCl2, 1 μ mol/L ZnCl2 )中浸泡3 h, 在孵育液 (不含Triton X-100的洗脱液)中37 ℃孵育42 h , 用染色液 [0.05%(质量分数)考马斯亮蓝R250, 30%(体积分数)甲醇, 10%(体积分数)乙酸]染色1 h, 脱色液 (30%甲醇, 10%乙酸)脱色至在蓝色背景上出现透亮条带。用凝胶成像分析系统对明胶酶谱结果进行灰度扫描 。
先用消毒过的记号笔在六孔板背后, 用直尺比着均匀地划横线, 大约每隔0.5~1.0 cm一道, 横穿过孔。每孔至少穿过5条线。细胞转染48 h后, 用Tip头比着直尺, 尽量垂至于背后的横线划痕, Tip头要垂直, 不能倾斜(划痕时细胞要密一些, 尽量长满)。用PBS洗细胞3次, 去除划下的细胞, 每孔加入fibronectin (16 mg/L), 用含5%(体积分数)热灭活新鲜小牛血清的RPMI1640于37 ℃ 5%(体积分数) 的CO2孵育箱中继续培养。第0、12和24小时取样, 倒置显微镜下观察照相, 并计算其划痕修复率或迁移细胞数。每组均设3个平行样本, 每个样本均观测2个视野(× 400), 取其平均值。划痕修复率=[(第0小时划痕宽度-第12或24小时划痕宽度)/第0小时划痕宽度]× 100%。
趋化因子的制备:处于对数生长期的 NIH3T3 细胞用无血清DMEM洗两次, 每瓶加入无血清的 DMEM培养液3 mL, 24~48 h后离心收集上清, 4 ℃ 12 000 r/min离心15 min, 取上清备用。在 Boyden 小室的下室加入该上清 200 μ L作为趋化因子; 上下室之间铺直径为 13 mm、孔径为8 μ m聚碳酸酯微孔滤膜, Fileter光面朝上。膜上均匀铺50 μ L用无血清DMEM 稀释的人工基底膜胶 Matrigel; 小室置于37 ℃温箱中 30 min, 使Matrigel充分聚合。各组细胞转染48 h, 消化后进行活细胞计数, 用含10%(体积分数)血清的1640培养液将细胞浓度调整至5× 105个/mL。在上室中贴壁匀速加入含 2× 105个细胞的细胞悬液400 μ L, 37 ℃ 5% (体积分数)CO2孵育箱中放置6~12 h; 吸去上室液体, 湿棉签擦去膜表面未侵袭的细胞, 生理盐水漂洗, 甲醇固定30 min, 常规 HE染色; 每组设3个平行样本, 每张膜计数5个200倍显微镜视野下的穿膜细胞数, 取其平均值, 进行统计学分析(200倍光镜下将膜用水平线及垂直线分为四个象限, 在象限及膜中心各选取1个视野计数, 取其平均值)。
载玻片培养细胞至 60%~70%融合, PBS洗涤, 4 ℃预冷的丙酮固定20 min, PBS洗涤5 min/次, 共3次。10%(体积分数)马血清封闭室温1 h(勿洗涤), 直接滴加TMSG-1羊抗人多克隆抗体和ATP6V0C兔抗人多克隆抗体的混和液[TMSG-1为1 ∶ 100(体积比)稀释, ATP6V0C为1 ∶ 250(体积比)稀释], 湿盒中4 ℃过夜孵育或室温孵育1~2 h。 PBS洗涤10 min/次, 共3 次, 滴加 FITC标记兔抗山羊IgG和 TRITC标记山羊抗兔IgG的二抗混合液[两种抗体均为1 ∶ 200(体积比)稀释], 室温避光孵育1 h。PBS洗涤5 min/次, 共3次, DAPI染核, 室温避光3 min。PBS洗涤5 min/次, 1次, 甘油/PBS体积比 9 ∶ 1封片, 晾干。荧光显微镜下观察。
采用 Bio-Rad 图像处理系统 ChemiDox XRS成像系统采集凝胶条带, 用quantity-One图像分析软件进行条带灰度扫描。
应用SPSS 13.0统计软件对数据进行统计学分析, 计量资料以均数± 标准差表示, 3组数据若方差齐则选用单因素方差分析, 其两两比较采用LSD-t检验; 3组数据若方差不齐, 则进行方差不齐的校正t检验。P< 0.05认为差异具有统计学意义。
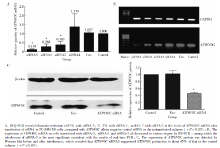
采用RT-PCR法显示ATP6V0C mRNA在不同转移潜能前列腺癌细胞系中均有表达, 但在高转移潜能PC-3M-1E8(0.370± 0.023)和PC-3M(0.195± 0.011)细胞系中的表达高于在低转移潜能细胞系PC-3M-2B4(0.054± 0.009)和PC-3(0.091± 0.005)中的表达, 两两比较结果显示, 任意两组间差异均有统计学意义(P< 0.05, 图1A)。
采用荧光real-time RT-PCR法检测ATP6V0C 基因在前列腺癌细胞系中的表达。ATP6V0C扩增后的片段长度约为118 bp, GAPDH扩增后的长度片段约为186 bp, 根据2-Δ Δ CT计算, ATP6V0C mRNA在PC-3M-1E8和PC-3M(高转移潜能)细胞系中的表达明显高于在PC-3M-2B4和PC-3(低转移潜能)细胞系中的表达, 其中ATP6V0C在PC-3M-1E8中的表达量最高, 差异具有统计学意义(P< 0.05, n=3, 图1B)。
采用Western blot法检测ATP6V0C在不同转移潜能前列腺癌细胞系中的表达, 结果显示ATP6V0C蛋白在高转移潜能细胞系PC-3M-1E8(0.481± 0.097)和PC-3M中的表达(0.333± 0.088)高于在低转移潜能细胞系PC-3M-2B4(0.115± 0.037)和PC-3(0.128± 0.032)中的表达, 差异具有统计学意义(P=0.002, 图1C和1D)。
 | 图1 不同转移潜能前列腺癌细胞系中ATP6V0C的表达Figure 1 ATP6V0C expression in prostate cancer cell lines |
荧光real-time RT-PCR结果显示, siRNA-3、siRNA-1和siRNA-2对ATP6V0C mRNA的沉默效率分别为87%、71.2%和61.7%, 与空白对照组(control)及阴性对照组(Unc)相比, 差异有统计学意义 (P< 0.05, 图2A)。再次用real-time RT-PCR进行验证, 发现转染siRNA-3、siRNA-1和siRNA-2的细胞中ATP6V0C mRNA的表达均有不同程度的降低, 其中siRNA-3的干扰效果最明显(图2B), 基本与real-time RT-PCR的结果吻合, 所以本实验选取siRNA-3为有效的siRNA用于后续实验。Western blot结果显示, siRNA-3干扰细胞后ATP6V0C蛋白的表达较空白对照组及无关阴性对照组相比显著下降(P< 0.05, 图2C)。
 | 图2 筛选具有较好沉默效率的ATP6V0C siRNAFigure 2 Effects of ATP6V0C siRNAS on the expression of ATP6V0C in PC-3M-1E8 cells |
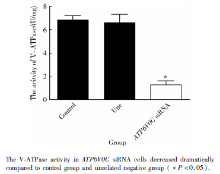
采用GEMEND公司的V-ATPase活性检测试剂盒来检测细胞内V-ATPase的活性, 严格按照GEMEND试剂盒步骤来检测3组细胞的V-ATPase活性, 计算公式:V-ATPase活性=(样品D差值-背景D差值)÷ (浓度× 31.1)× 103单位/mg, 该实验重复3次。空白对照组、阴性对照组和干扰组细胞的V-ATPase活性分别为6.867± 0.306、6.583± 0.705、1.297± 0.342, 与空白对照组及阴性对照组相比, 干扰组细胞内的V-ATPase的活性明显下降(P< 0.05, 图3)。
 | 图3 ATP6V0C siRNA可抑制细胞的V-ATPase活性Figure 3 siRNA against ATP6V0C decrease the activity of V-ATPase in PC-3M-1E8 cells |
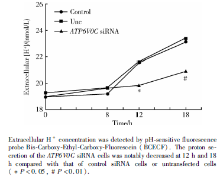
采用BCECF氢离子荧光探针检测细胞外H+浓度, 绘制标准pH值曲线。ATP6V0C siRNA转染组细胞外H+浓度在第12和18小时均低于空白对照组及阴性对照组 (P< 0.05, 图4) 。
 | 图4 ATP6V0C siRNA可降低细胞外H+浓度Figure 4 ATP6V0C siRNA decrease extracellular H+concentration in PC-3M-1E8 cells |
siRNA转染组MMP-2蛋白在细胞内的表达和上清液中的分泌与空白对照组及阴性对照组相比, 差异无统计学意义(P> 0.05, 图5A), 且3组细胞内MMP-9蛋白的表达, 差异也无统计学意义 (P> 0.05, 图5B), 说明ATP6V0C表达下调后可能并没有影响MMP-2及MMP-9的表达和分泌(MMP-9的分泌未检测出)。
 | 图5 ATP6V0C siRNA对细胞中MMP-2和MMP-9蛋白的表达分泌无影响, 但能抑制细胞培养上清液中MMP-9的活性Figure 5 The effect of ATP6V0C siRNA on the expression, secretion and activity of MMP-2 and MMP-9 in the culture cells |
采用明胶酶谱法检测细胞培养上清液中MMP-9的活性, 与空白对照组及阴性对照组相比, ATP6V0C siRNA转染组的细胞上清液中MMP-9的活性明显减弱(P< 0.01, 图5C, MMP-2的活性未检测出)。
采用细胞划痕修复实验来检测ATP6V0C的siRNA对PC-3M-1E8细胞迁移能力的影响。在划痕修复实验中, 3组细胞均设3个平行样本, 每个样本观测2个视野 (× 400), 取其平均值, 24 h后空白对照组、阴性对照组及ATP6V0C siRNA转染组细胞的划痕修复率分别为47.00%± 1.00%、50.67%± 1.45%和22.33%± 1.20%, 差异具有统计学意义 (P< 0.05), 并且转染组细胞迁移的个数较其他两组也明显减少(图6), 说明下调ATP6V0C的表达后抑制了PC-3M-1E8细胞的体外迁移能力。
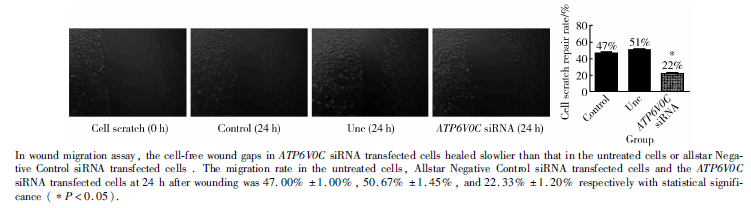 | 图6 ATP6V0C siRNA抑制细胞体外迁移能力Figure 6 siRNA targeting ATP6V0C weakened the migration of PC-3M-1E8 cells |
在侵袭实验中, 3组细胞均设3个平行样本, 每个样本观测5个视野(× 200), 空白对照组、阴性对照组及ATP6V0C siRNA转染组发生侵袭的细胞个数分别为 67.67± 2.186、71.00± 3.055和38.33± 1.202, 差异具有统计学意义 (P< 0.05, 图7), 说明下调ATP6V0C的表达后抑制了PC-3M-1E8细胞的体外侵袭能力。
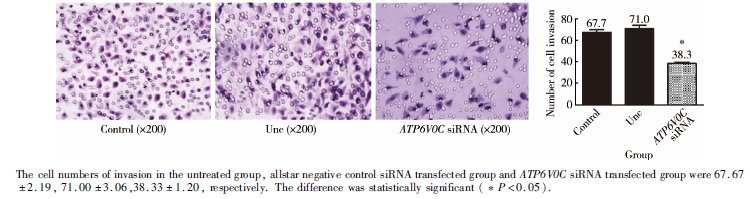 | 图7 ATP6V0C siRNA抑制PC-3M-1E8细胞的体外侵袭能力Figure 7 Using boyden chamber invasion assay, the ATP6V0C siRNA transfected cells displayed dramatically decreased invasion ability when compared with the untreated cells or allstar negative control siRNA transfected cells |
RT-PCR结果显示, 转染siRNA ATP6V0C后, PC-3M-1E8细胞中LASS2/TMSG1 mRNA的表达较空白对照组及阴性对照组分别减少77%和74%(P< 0.05, 图8A)。real-time RT-PCR也得出同样的结果, 当ATP6V0C基因被干扰后, LASS2/TMSG1 mRNA的表达也受到明显抑制, 与空白对照组及阴性对照组相比, 差异具有统计学意义(P< 0.05, 图8B) 。
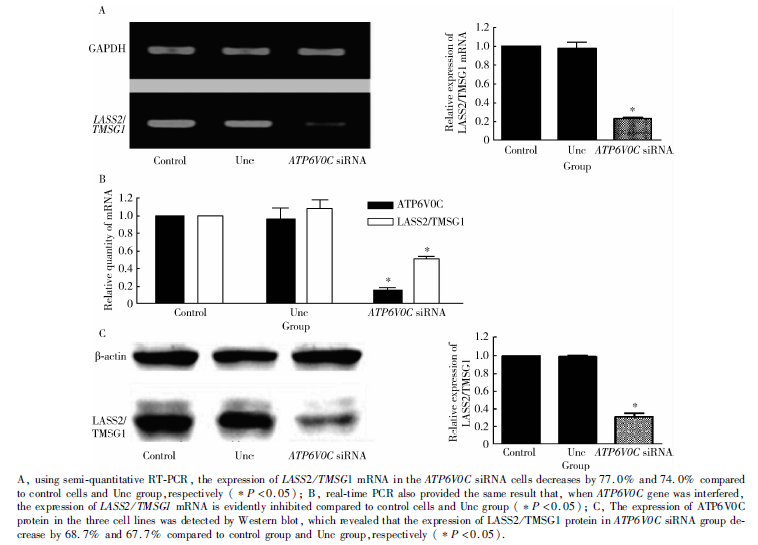 | 图8 ATP6V0C siRNA抑制PC-3M-1E8细胞中LASS2/TMSG1的表达Figure 8 ATP6V0C siRNA inhibited the expression of LASS2/TMSG1 in PC-3M-1E8 cells |
Western blot检测, 靶向ATP6V0C siRNA转染组LASS2/TMSG1蛋白的表达较空白对照组及阴性对照组分别减少了68.7%和67.7%(P< 0.05, 图8C)。
利用激光共聚焦显微镜观察免疫荧光双重染色, 发现LASS2/TMSG1蛋白(绿色)和ATP6V0C蛋白(红色)主要都在胞浆中表达, 且两者在胞浆中存在共定位(黄色)。PC-3M-1E8细胞转染ATP6V0C siRNA后, 干扰组与空白对照组及阴性对照组相比, 胞浆中LASS2/TMSG1和ATP6V0C的表达均明显下降, 且两种蛋白共定位信号明显减弱(图9)。
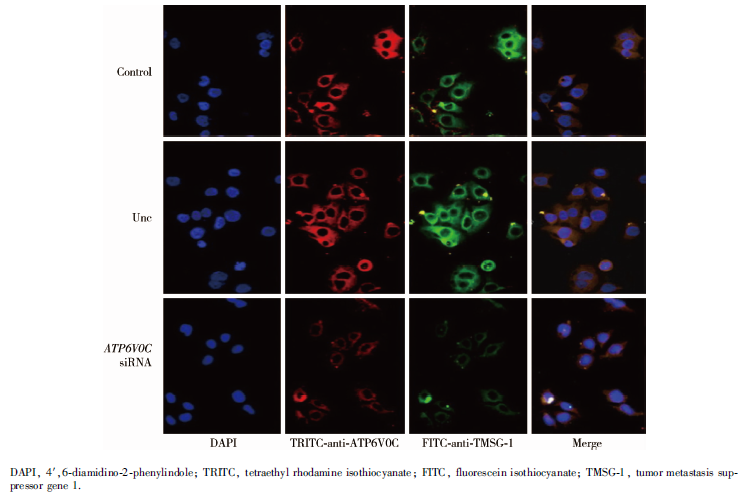 | 图9 免疫荧光双重染色直接观察LASS2/TMSG1和V-ATPase之间的相互作用。利用激光共聚焦显微镜观察免疫荧光双重染色, 结果显示, LASS2/TMSG1蛋白(绿色)和ATP6V0C蛋白(红色)主要都在胞浆中表达, 且两者在胞浆中存在共定位(黄色)。PC-3M-1E8细胞转染ATP6V0C siRNA后, 干扰组与空白对照组及阴性对照组相比, 胞浆中LASS2/TMSG1和ATP6V0C的表达均明显下降, 且两种蛋白共定位信号明显减弱Figure 9 Double immunofluorescent staining confirmed the interaction between ATP6V0C and LASS2/TMSG1, showing that LASS2/TMSG1 protein(green) and ATP6V0C protein (red) are mainly co-localized in plasma (yellow). After the PC-3M-1E8 cells are transfected with ATP6V0C siRNA, expression of LASS2/TMSG1 and ATP6V0C both decreased significantly compared to control group and negative group, and the co-localization signal was decreased evidently |
肿瘤组织中, ATP6V0C能够泵出质子导致细胞外酸化, 有助于维持相对碱性的细胞质环境和相对酸性的细胞外环境, 而细胞外的低pH值, 可通过激活蛋白水解酶促进细胞外基质(extracellular ma-trixc, ECM)的降解和重塑, 从而促进癌细胞的侵袭和转移[10, 11]。V-ATPase除了通过调节pH值的机制来影响肿瘤的侵袭转移外, 还可以通过其他机制来改变细胞的黏附及迁移过程, 从而促进肿瘤细胞的浸润。ATP6V0C可以与整合素直接作用, 影响整合素在细胞膜上分布的浓度及在膜系统上的循环, 改变细胞的黏附性[12]。V-ATPase V1结构域的C亚基还通过和actin锚定而与细胞骨架相连[13], 同时, V-ATPase的B亚基氨基端也有与actin的结合点[14], V-ATPase与细胞骨架相连从而影响细胞的迁徙过程, 此外, V-ATPase的c亚基ATP6V0C本身也有血小板源性生长因子和乳头瘤状病毒属E5蛋白等结合部位, 而这些物质都参与调控了肿瘤细胞的增殖及黏附过程[8, 15, 16]。
本实验采用了半定量RT-PCR、荧光real-time RT-PCR和Western blot等方法检测了不同转移潜能前列腺癌细胞系中V-ATPase的c亚基ATP6V0C的mRNA及蛋白表达情况, 发现在高转移潜能细胞系PC-3M-1E8和PC-3M中ATP6V0C的mRNA及蛋白表达均明显高于在低转移潜能细胞系PC-3M-2B4和PC-3中的表达, 其中ATP6V0C在PC-3M-1E8细胞中的表达量最高, 这再一次证明了ATP6V0C是一种肿瘤转移相关基因, 在肿瘤侵袭转移过程中发挥了重要的作用, 随后本研究选择了PC-3M-1E8细胞进行ATP6V0C基因沉默实验。
本研究设计并合成了特异性沉默V-ATPase c亚基ATP6V0C基因的siRNA, 下调人前列腺癌高转移潜能细胞系PC-3M-1E8中ATP6V0C的表达, 进一步探讨ATP6V0C在肿瘤侵袭和转移中的作用及其分子机制, 采用GEMEND公司V-ATPase活性检测试剂盒直接检测肿瘤细胞内V-ATPase的活性, 并发现转染特异性ATP6V0C的 siRNA后, 肿瘤细胞中V-ATPase的活性明显下降, 同时通过BCECF氢离子敏感荧光探针检测发现, 转染后细胞外H+浓度明显降低, 提示沉默ATP6V0C可抑制V-ATPase的活性, 从而抑制了该质子泵跨膜运输H+的功能。本研究还采用GEMEND公司明胶酶谱试剂盒检测发现, 细胞上清液中MMP-9的活性明显减弱, 这可能与质子泵向胞外泵出的H+减少有关。因为细胞外的高pH值抑制了对H+敏感的蛋白水解酶类如MMP家族(MMP-9等)的激活[17], 从而阻碍了细胞外基质(extracellular matrix, ECM)的降解和重塑, 抑制了肿瘤细胞的侵袭和转移。细胞划痕修复实验及transwell体外侵袭实验均可证明, 当ATP6V0C的表达被沉默后, PC-3M-1E8细胞的体外迁移能力和侵袭能力都明显减弱了。
LASS2/TMSG1基因是北京大学基础医学院病理学系于1999年首先发现的一种新的肿瘤转移抑制基因[18], 在GenBank中的登记号为AF189062, 并陆续得到其他实验室的证实。2000年2月, 日本东京大学实验室从人10周胚胎cDNA文库中克隆出一新基因, 登记序列AK001105, 与TMSG1氨基酸序列有87%的同源性。2001年7月, 美国NIH癌症研究所从人类皮肤黑色素瘤中克隆出一新基因[19], 登记序列BC010032, 与LASS2/TMSG1有高度同源性。2001年9月, 上海复旦大学遗传研究所应用5'-RACE技术从人肝cDNA文库中克隆出一与酵母长寿保障基因LAG1高度同源的新基因, 命名为LASS2(homo sapiens longevity assurance homologue 2 of yeast LAG1), 与TMSG1具有100%同源性[6, 7]。2008年, 以色列人发现LASS2/TMSG1具有利用长链脂肪酰辅酶A(C20~C26)合成神经酰胺的功能[6, 7], 并将其更名为神经酰胺合成酶2(ceramide synthase 2, CerS2), 因此, LASS2/TMSG1基因的同义词还包括SP260基因和CerS2基因等。LASS2/TMSG1在低转移潜能的前列腺癌细胞系中高表达, 而在高转移潜能的前列腺癌细胞系中低表达[19]。2002年, 潘辉等[7]采用酵母双杂交和GST pull-down实验发现LASS2(即LASS2/TMSG1)蛋白可以和V-ATPase的c亚基ATP6V0C直接结合。2007年, 谭宁等[9]发现LASS2/TMSG1基因对肝癌细胞HCCLM3的转移具有显著抑制作用, 推测其作用机制可能是因为LASS2/TMSG1结合ATP6V0C后抑制了V-ATPase的活性, 从而抑制肿瘤细胞的生长、侵袭和转移。2010年, 本课题组证实采用基因干扰的方法下调LASS2/TMSG1的表达[20], 能够增加细胞外氢离子浓度, 增强V-ATPase的活性, 并抑制肿瘤细胞的凋亡, 促进前列腺癌的侵袭和转移。目前研究主要是针对LASS2/TMSG1对V-ATPase活性的作用, V-ATPase是否对LASS2/TMSG1具有反馈调节作用尚未见研究报道。
本实验采用短片段双链siRNA来干扰ATP6VOC基因在人前列腺癌高转移细胞系PC-3M-1E8(ATP6V0C高表达)中的表达, 研究其对LASS2/TMSG1 mRNA和蛋白表达量的影响。采用RT-PCR、real-time PCR和Western blot等方法证明, 沉默ATP6V0C后, 干扰组LASS2/TMSG1 mRNA和蛋白表达均明显低于空白对照组和阴性对照组; 本研究还采用免疫荧光双重染色的方法来直接观察干扰后LASS2/TMSG1和ATP6V0C之间的相互作用, 发现干扰后两种蛋白在胞浆内的表达及其共定位信号均明显减弱, 提示ATP6V0C对LASS2/TMSG1可能具有反馈调节作用。
目前大量工作都是针对LASS2/TMSG1基因的功能性研究, 但是对其上游事件(基因表达调控)和下游事件的研究甚少见。通过生物信息学分析发现, 人类LASS2/TMSG1蛋白即LASS2蛋白是由homeodomain和TLC结构域组成, 其中homeodomain是DNA结合区域, 包含60个氨基酸, 可以与特定的DNA序列识别、结合, 具有转录因子的功能, 有研究报道[21], homeodomain与真核生物发育过程中的转录调控有关。2012年, 本课题组构建了LASS2/TMSG1基因及其截短体的质粒并转染PC-3M-1E8细胞, 发现LASS2/TMSG1主要也是通过其homedomain与V-ATPase的c亚基相互作用[22]。虽然LASS2/TMSG1拥有转录因子功能的homeodomain, 但是目前仍然没有确切证实LASS2/TMSG1蛋白是一种潜在的转录因子[23, 24]。2011年, 本课题组通过蛋白质免疫共沉淀、染色质免疫共沉淀和凝胶迁移阻滞等实验证实了转录因子KLF6与Sp1共同作用于LASS2/TMSG1基因第一外显子内的调控序列, 从而参与LASS2/TMSG1的转录激活; 同时也发现在LASS2/TMSG1基因的转录起始点上游存在多个抑制性调控区域, 鉴定这些抑制性顺式作用原件所结合的重要的反式作用因子尚待进一步探索。本研究利用siRNA干扰ATP6V0C的同时, 检测到LASS2/TMSG1的表达水平亦明显下降。这种正性调节作用可能是由于下调ATP6V0C的表达后, 通过某种途径激活了与LASS2/TMSG1上游抑制性调控区域结合的转录因子, 从而抑制了LASS2/TMSG1基因的表达, 但目前对V-ATPase参与的信号通路及LASS2/TMSG1在细胞内的转录调控机制尚不清楚, 后期还需要进行大量实验来完善这些工作, 并争取将LASS2/TMSG1和V-ATPase的检测应用于临床中, 为判断肿瘤侵袭、转移及预后提供新的分子生物学标志物及药物治疗的新靶点。
综上所述, 特异性siRNA沉默ATP6V0C基因后能抑制人前列腺癌细胞的体外迁移及侵袭能力, 其机制与沉默ATP6V0C后抑制了V-ATPase的活性, 使肿瘤细胞外氢离子浓度降低, 进而影响MMP-9等基质金属蛋白酶的激活, 并阻碍ECM的降解和重构有关, 并且反向提示V-ATPase的异常导致的H+浓度的变化是肿瘤细胞扩散、恶变的初始条件和促进因子之一, 因此, 通过各种途径抑制V-ATPase的活性, 尤其是下调其c亚基ATP6V0C的表达来达到抑制肿瘤生长扩散的目的是可行的。此外, LASS2/TMSG1能够与V-ATPase的c亚基ATP6V0C结合, 影响V-ATPase的活性; 反之, 用特异性siRNA沉默ATP6V0C基因的表达, 也能够抑制LASS2/TMSG1基因的表达, 说明ATP6V0C对LASS2/TMSG1具有反馈调节作用, 其机制可能与ATP6V0C的下调通过某种途径激活了与LASS2/TMSG1上游抑制性调控区域结合的转录因子有关。
The authors have declared that no competing interests exist.
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|