

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
肥胖者唾液微生物宏基因组学特点
[吴宇佳1, 2 , 迟晓培2 , 陈峰3 , 邓旭亮1, △  ]
]
]
|
|


目的 探讨肥胖者口腔唾液微生物的特点,比较肥胖者与正常体重者口腔唾液微生物组成、基因功能及代谢通路上的差异。方法 研究纳入无全身系统性疾病、无牙周炎及口腔黏膜病的肥胖者及性别、年龄相匹配的正常体重者,收集研究对象的非刺激性唾液。提取唾液样本DNA,用高通量测序方法进行宏基因组分析,结果经数据质控后进行物种分类及注释,差异物种线性判别分析(linear discriminant analysis, LDA),基因预测,基因集构建和功能注释分析。结果 过滤后每个样本得到可分类的、细菌的DNA序列平均条数为2 630 428。唾液微生物群落共包括 11个菌门、19个菌纲、26个菌目、41个菌科、62个菌属和164菌种。肥胖组与正常体重组微生物群落的优势菌门相同,为变形菌门、厚壁菌门、拟杆菌门、放线菌门和梭杆菌门,两组在纲、目、科、属、种等分类水平上均发现相对丰度差异具有统计学意义的物种。在纲水平上,Negativicutes纲和丹毒丝菌纲(Erysipelotrichia)丰度在肥胖组中的相对丰度高于正常体重组( P<0.05),黄杆菌纲(Flavobacteriia)和Bateroidetes纲在肥胖组中的相对丰度低于正常体重组( P<0.05)。9个菌属在两组间丰度差异具有统计学意义;种水平上,16个菌种的丰度差异具有统计学意义,其中产黑色素普雷沃氏菌( Prevotella melaninogenica)、唾液普雷沃氏菌( Prevotella salivae)、 Solobacterium moorei和极小阿托波氏菌( Atopobium parvulum)等在肥胖组中相对丰度高于正常体重组( P<0.05),而血链球菌( Streptococcus sanguinis)在正常体重组中相对丰度高于肥胖组( P<0.05)。对基因预测结果进行分析,发现肥胖组样本基因数目较正常体重组多,差异有统计学意义( P<0.05),其中与营养和能量代谢、环境信息处理、人体疾病等通路相关的基因在肥胖组唾液样本中显著富集( P<0.01)。结论 肥胖者与正常体重者的口腔唾液微生物在物种组成、基因数目及代谢通路上均存在差异,值得今后扩大样本量进一步研究。
Objective: To investigate the characterization of the salivary microbiome in people with obesity and the differences in microbial composition, gene function and metabolic pathways of salivary microbiome between people with obesity and normal weight controls.Methods: The study was carried out in people with obesity and age- and sex-matched normal weight controls. None of these selected participants had the systemic disease, oral mucosal disease or periodontal disease. Unstimulated saliva samples were collected and oral examination was conducted. DNAs from saliva samples were extracted and sequenced in an Illumina NextSeq 500 platform. Community composition, linear discriminant analysis of taxonomic differences,gene prediction, gene set construction and annotation of gene function were performed.Results: The classified bacterial reads of the samples were 2,630,428 for each sample. A total of 11 phyla, 19 classes, 26 orders, 41 families, 62 genera and 164 species were detected ultimately. All samples had the same predominant phyla (Proteobacteria, Firmicutes, Bacteroidetes, Actinobacteria and Fusobacteria). There were statistical differences between the groups at the class, order, family, genus and species levels. At the class level, Negativicutes and Erysipelotrichia were more abundant in the obesity group, while Flavobacteriia and Bateroidetes dominated in normal weight group ( P<0.05). At the species level, 16 showed significant differences in relative abundance among the groups, in which Prevotella melaninogenica, Prevotella salivae, Solobacterium moorei and Atopobium parvulum ware more abundant in the obesity group, whereas Streptococcus sanguinis dominated in normal weight group ( P<0.05). The people with obesity had a higher number of salivary microbial genes ( P<0.05). We produced statistics on gene prediction and found salivary microbiome of obesity group had a higher number of genes ( P < 0.05). Genes associated with the pathways of metabolism and environmental information processing and human diseases were significantly enriched in the saliva samples of people with obesity ( P < 0.01).Conclusion: Significant differences were seen in composition, gene function and metabolic pathways of salivary microbiome between people with obesity and normal weight people. We hope to go on further study with larger sample size in the near future.
口腔是人体重要的微生态系统之一, 口腔内定植着复杂的微生物群落, 这些微生物广泛参与了宿主的代谢、营养、免疫等生理活动。口腔微生物群落组成和结构的变化, 影响着口腔微生态的平衡, 对口腔健康或疾病有重要作用。口腔作为消化道的起始部分, 是微生物进入人体的门户之一, 口腔微生物同时还与全身系统性疾病相关。通过二代测序技术研究发现, 口腔微生物与口腔鳞状细胞癌、炎症性肠病等疾病相关[1, 2]。近年来, 新兴的宏基因组学研究方法以环境样本中的微生物群体基因组为研究对象, 直接从环境样本中提取全部微生物的DNA, 构建宏基因组文库, 利用基因组学的研究策略分析环境样本所包含的全部微生物的基因组成、群落功能及参与的代谢通路[3]。宏基因组学研究不仅从整体上揭示了健康人体口腔微生物的生物多样性, 还在全身疾病(如肝硬化[4]、类风湿性关节炎[5])患者的肠道中检测到了口腔来源的微生物, 进一步提示了口腔微生物与一些全身系统性疾病存在密切关联。
肥胖是一种多因素的慢性代谢性疾病, 具有患病率高, 患病人数增长速度快等特点, 已成为威胁人类健康的重要公共卫生问题之一[6, 7]。近二十年来, 随着居民生活水平的改善和生活方式的转变, 中国人肥胖的患病率逐年增长, 最新调查报告显示中国目前成人肥胖人口数超越美国, 成为全球肥胖人口最多的国家[8]。已有大量动物模型分析与人体试验表明, 肥胖与肠道菌群的变化密切相关[9, 10]。人体口腔与肠道微生物可能存在结构和功能上的重叠, 同一个体的口腔微生物群落类型与其肠道微生物群落类型显著相关[11]。应用人类口腔微生物鉴定芯片(human oral microbe identification microarray, HOMIM)和16SrRNA测序技术发现口腔内微生物的种类和数量可能与肥胖相关[12, 13], 但由于检测精度和分辨率有限, 未能展现肥胖者口腔微生物全貌, 此外, 口腔微生物功能基因及代谢通路与肥胖的关系目前尚未见报道。本研究旨在应用宏基因组学方法分析比较无牙周炎的肥胖者与健康体重者唾液微生物的组成、基因功能及代谢通路之间的差异, 为更深入研究口腔菌群与肥胖之间的关联提供线索。
选取北京大学口腔医院自2016年6月至9月在门诊进行常规口腔检查牙周健康的患者共6名, 包括正常体重者3名, 肥胖者3名作为研究对象。受试者的纳入及排除标准参考以往报道[12]。
纳入标准:(1) 年龄 ≥ 18岁; (2) 肥胖者身体质量指数(body mass index, BMI)≥ 30 kg/m2, 正常体重者 18.5 ≤ BMI < 24 kg/m2; (3)口腔健康状况:全口牙平均探诊深度 ≤ 3 mm, 无附着丧失, 牙龈出血指数 ≥ 3的牙数目不超过全口牙数的10%, 口内至少有20颗自然非龋牙。
排除标准:(1)有全身系统性疾病(如糖尿病、高血压、高血脂或胃肠道疾病等)者; (2)6个月内接受过肥胖相关治疗者; (3)3个月内接受过牙周治疗或服用过抗生素者; (4)正接受正畸治疗者; (5)吸烟者。
本研究开始前获得北京大学口腔医院伦理委员会审查批准(PKUSSIRB-201627023), 所有研究对象包括肥胖者和正常体重者均签署知情同意书。
唾液采集方法参考美国人类微生物组计划中唾液样本采集的标准流程[14], 并稍作改良。受试者在唾液样本收集前一天的晚餐后不进食, 睡前刷牙; 收集样本当天清晨不进食、不饮水、不刷牙。取样时间在上午8时至11时, 要求受试者保持坐位, 手持一个50 mL无菌离心管, 保持唾液在口中持续至少1 min, 然后使唾液自然流出到离心管中, 重复多次直到收集的唾液达到3 mL, 将离心管即刻置于冰上。 1 h内将样本送至实验室12 000× g离心10 min, 留取沉淀并分装后-80 ℃冰箱冻存。
所有受试者填写调查问卷, 内容包括基本资料、全身健康情况及口腔卫生习惯, 由一名牙周科医师对受试者进行口腔检查, 并记录口腔状况, 其中包括龋失补指数(decayed, missed, and filled teeth index, DMFT)和牙周袋探诊深度(periodontal pocket depth, PPD)。
唾液样本DNA提取使用DNA提取试剂盒(QIAamp DNA Blood Mini Kit, 德国Qiangen公司), 按说明书步骤, 经过微生物细胞破碎、DNA抽提、纯化, 提取唾液微生物DNA。使用分光光度计(NanoDrop 8000, 美国Thermo Scientific公司)检测DNA样品的纯度, 凝胶电泳进行质检, 使用Qubit 2.0荧光计(美国Invitrogen公司)检测基因组DNA浓度。
将提取的宏基因组DNA打断成约500 bp的片段后, 按照建库试剂盒(NEBNext Ultra DNA Library Prep Kit, 美国Illumina公司)的说明书操作进行文库构建。DNA文库构建完成后, 采用Nextseq 500高通量测序平台(美国Illumina公司)进行宏基因组高通量测序。
用MOCAT2软件对原始数据进行质量控制, 去除低质量序列和重复片段, 并通过比对人类参考基因组Hg19剔除人源DNA序列, 获得高质量序列数据; 使用MEGAN 5 软件和MetaPhlAn软件进行菌群组成分析, 获得菌群相对丰度, 并用GraPhlAn 0.97软件构建菌群分类树; 使用SOAPdenovo 1.0.5对每个样本的高质量序列数据进行从头组装, 获得scaf-tig序列, 并用MetaGeneMark 2.8软件对scaftig序列进行基因预测, 获得预测的基因集, 然后聚类去冗余构建非冗余基因集; 用MOCAT2中的比对工具将高质量序列比对到非冗余基因集上, 获得基因的相对丰度; 使用DIAMOND软件将非冗余基因集比对到京都基因与基因百科全书(Kyoto Encyclopedia of Genes and Genomes, KEGG)数据库, 使用HUMAnN的核心算法, 统计获得每个基因的KEGG orthology(KO)注释信息, KO的相对丰度通过累加所注释上的基因相对丰度获得[15, 16, 17, 18, 19, 20, 21, 22, 23]。
受试者基本信息采用统计软件SPSS 22.0来计算均数和标准误, 并进行独立样本t检验。组间差异物种使用LEfSe软件进行线型判别分析(linear discriminant analysis, LDA), 将具有显著性差异的物种标注在聚类树的不同分支上, 两组差异物种的显著作用使用LDA分值绘制的柱形图表示。通过与KEGG数据库进行比对, 对序列进行功能注释, 并采用Kruskal-Wallis秩和检验进行分析。
研究对象共6名, 包括3名肥胖者及3名性别、年龄相匹配的正常体重者。肥胖组与正常体重组年龄、口腔健康情况包括龋失补指数和平均牙周探诊深度的差异均无统计学意义(P > 0.05)。两组间BMI有明显统计学差异[(19.9 ± 0.43) kg/m2vs.( 34.4 ± 1.46) kg/m2, P = 0.0003]。所有受试者基本信息见表1。
| 表1 受试者基本信息表 Table 1 Demographic and clinical characteristics |
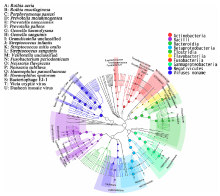
用高通量测序方法对样本中提取到的总DNA进行双末端测序, 得到基因组序列。进行数据质量控制后, 共得到28.2 Gb数据, 可分类的、细菌的DNA序列每个样本平均条数为2 630 428。对样本测得的数据进行物种分析并绘制物种组成分类树(图1)。唾液微生物宏基因数据在界水平可以注释到细菌、真菌、病毒、古细菌4大类, 在门、纲、目、科、属、种水平分别有11、19、26、41、62、164个分类, 结果显示了在不同分类水平所包含的物种, 图1圆圈的10种颜色分别代表在分类树里面相对丰度最高的10个物种分支, 圆圈的面积越大表明其中包含序列越多。
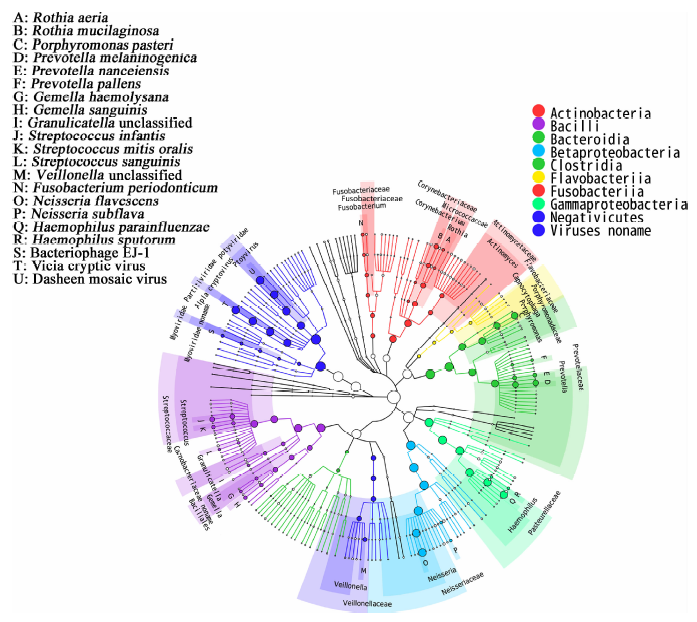 | 图1 1 物种组成分类树分析Figure 1 Phylogenetic tree analysis of the samples |
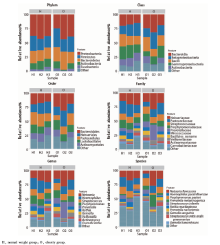
在门、纲、目、科、属和种水平分别对各唾液样本的菌种分布进行了展示(图2), 在门水平, 唾液菌群包含11个门, 其中所有样品的共有优势菌门为变形菌门(Proteobacteria)、厚壁菌门(Firmicutes)、拟杆菌门(Bacteroidetes)、放线菌门(Actinobacteria)和梭杆菌门(Fusobacteria), 其余6个门的丰度仅占不超过1%。在属水平上, 奈瑟球菌属(Neisseria)、嗜血菌属(Haemophilus)、链球菌属(Streptococcus)、卟啉菌属(Porphyromonas)、普雷沃氏菌属(Prevotella)、罗氏菌属(Rothia)、兼性双球菌属(Gemella)、韦荣球菌属(Veillonella)、放线菌属(Actinomyces)和颗粒链球菌属(Granulicatella)在各样本中均有较高的丰度。
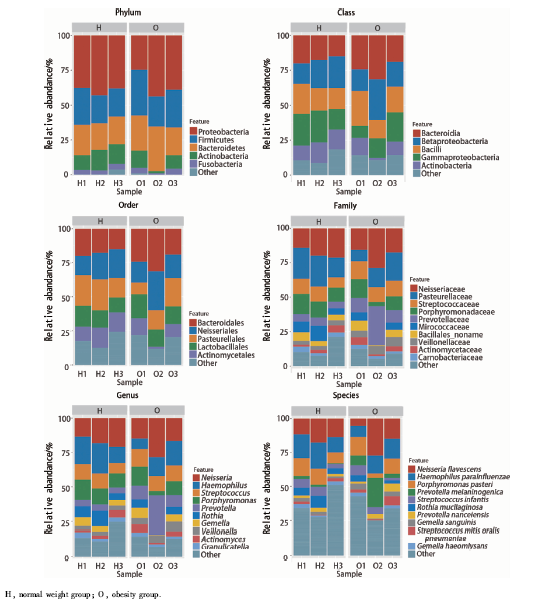 | 图2 各个唾液样本菌群在门、纲、目、科、属、种水平上的菌群组成Figure 2 Bacteria composition at phylum level, class level, order level, family level, genus level and species level of each saliva sample |
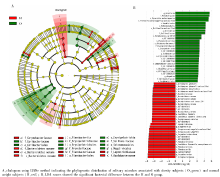
LEfSe分析发现在纲、目、科、属、种、株等水平两组唾液样本均存在相对丰度差异具有统计学意义(LDA score(log10)> 3)的物种(图3), 其中纲水平上差异物种有4种, Negativicutes纲和丹毒丝菌纲(Erysipelotrichia)丰度在肥胖组中较高, 而黄杆菌纲(Flavobacteriia)和Bacteroidetes纲丰度肥胖组则低于对照组。属水平差异存在统计学意义的物种有9种, 表现为普雷沃氏菌属(Prevotella)、韦荣球菌属(Veillonella)、Solobacterium属、阿托波氏菌属(Atopobium)在肥胖组中较高, 而棒状杆菌属(Corynebacterium)、二氧化碳嗜纤维菌(Capnocyto-phaga)属、拟普雷沃菌属(Alloprevotella)、Candidatus zinderia属和拟杆菌属(Bacteroidetes)丰度则在对照组中较高。本研究在种水平上比较肥胖者与正常体重者的唾液差异微生物, 发现16种菌的丰度差异存在统计学意义, 其中肥胖组的产黑色素普雷沃氏菌(Prevotella melaninogenica)、唾液普雷沃氏菌(Prevotella salivae)、Solobacterium moorei、极小阿托波氏菌(Atopobium parvulum)和殊异韦荣球菌(Veillonella dispar)的丰度高于正常体重组, 而血链球菌(Streptococcus sanguinis)在肥胖组的丰度明显低于正常体重组。
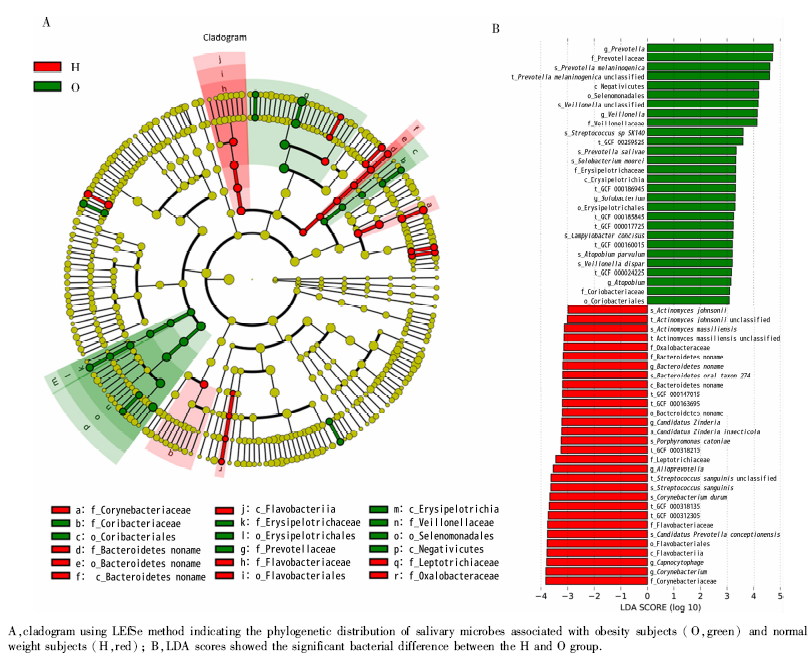 | 图3 正常体重组和肥胖组唾液微生物的、差异物种LEfSe分析及LDA分析Figure 3 LEfSe and LDA analysis of salivary microbiome between the H and O |
基因集构建的维恩图如图4A所示, 两组共有基因集包含84 002个基因, 肥胖组特有基因有18 854个, 正常体重组特有基因有6 807个。肥胖组基因集中的基因数目远高于正常体重组, 如图4B所示。
通过LEfSe软件分析得到两组间差异有统计学意义(P< 0.01, 校正后P< 0.1)的68个代谢通路, 绘制成热图结果如图5 所示。与营养和能量代谢、环境信息处理、人体疾病等通路相关的基因在肥胖组唾液样本中富集, 其中营养和能量代谢通路包括碳水化合物代谢、糖原合成和代谢、能量代谢、氨基酸代谢及辅酶和维生素代谢的功能通路丰度显著高于正常体重组。整体表现为肥胖组唾液微生物物质代谢与能量代谢较正常体重组更为活跃。本研究还发现参与细菌入侵上皮细胞代谢通路和扩张性心肌病发生相关通路在肥胖组唾液样本中较高。
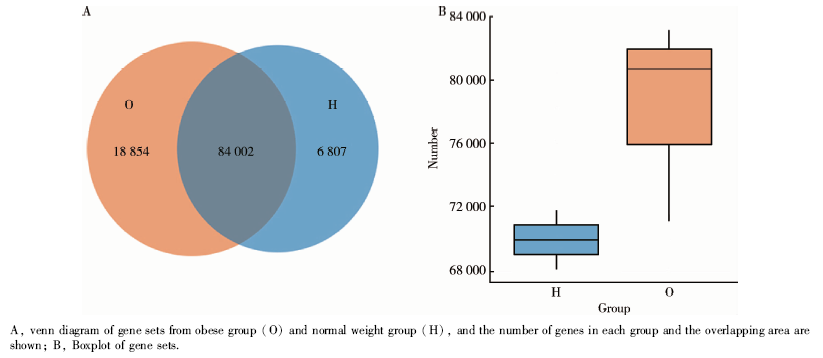 | 图4 正常体重组与肥胖组唾液微生物的基因集比较Figure 4 Comparisons of microbial gene sets in normal weight group and obese group |
 | 图5 正常体重组与肥胖组的差异功能通路的热图Figure 5 Difference of salivary microbiome KEGG pathway between the normal weight group and obese group |
本研究以严格的纳入标准, 选择无全身系统性疾病且牙周健康的人作为取样对象, 应用宏基因组学研究方法比较肥胖者和正常体重者唾液微生物组成、基因功能及代谢通路的差异。Goodson等[12]应用DNA探针的方法分析超重者及正常体重者唾液中40种细菌的数量, 首次提出口腔微生物和肥胖之间存在联系。Zeigler等[13]应用DNA-DNA棋盘杂交技术检测发现肥胖青少年龈下菌斑中细菌数量显著高于正常体重者, 但DNA-DNA棋盘杂交技术只能检测环境中有限的优势菌群, 检测精度不尽如人意。Piombino等[24]应用16S核糖体RNA高通量测序技术发现了男性肥胖者唾液微生物与正常体重者存在不同, 而其研究只精确到科水平, 不能全面反映唾液微生物的组成, 且无法在微生物基因功能和代谢通路层面进行解释。本研究采用宏基因组测序技术, 不仅能检测到已知序列的微生物, 还可发现未知微生物的基因序列, 从覆盖物种的广度和深度两方面丰富了样本微生物信息, 同时还对微生物的功能基因和代谢通路情况进行评价, 一定程度上揭示了肥胖者与正常体重者唾液菌群组成和基因功能的差异。
人体微生态系统包含肠道、口腔、鼻腔、皮肤、泌尿系统等几大组成部分, 大量研究表明, 人体各部位微生物群落组成和结构不同, 存在特征性微生物, 而在同一个体中, 各部位微生物在结构和功能上又存在一定联系[25]。在研究人体不同部位微生物群落时, Ding等[11]发现粪便样本与口腔样本微生物群落之间存在显著相关性(P=0.001), 而口腔样本中相关性最强是唾液样本, 普雷沃氏菌属在肠道和口腔内的相对丰度均较高, 且同一个体口腔和肠道内普雷沃氏菌属的相对丰度存在显著正相关。Zhang等[26]研究已证实肥胖者肠道中普雷沃氏菌相比对照组较多。本研究发现肥胖者唾液中普雷沃氏菌较正常体重者多, 与肠道研究结果相一致。普雷沃氏菌参与碳水化合物及蛋白质发酵, 产生乙酸盐和氢气。在肠道中, 普雷沃氏菌作为产氢菌与需氢的产甲烷菌互利共生, 使得机体从食物中更有效地摄取营养成分[27]。本研究发现在唾液微生物群落的功能通路水平上的显著差异, 且大多是肥胖组的表达高于正常体重组, 如碳水化合物代谢、糖原合成和代谢、能量代谢、氨基酸代谢及辅酶和维生素代谢等, 具体机制尚不明确, 仍需要加大样本量深入研究。
本研究还发现Solobacterium moorei和极小阿托波氏菌的丰度在肥胖组唾液样本中较高。Solobacterium moorei已被证实是口臭致病菌之一, 在口臭的发生发展中扮演重要角色[28], 极小阿托波氏菌与口臭同样密切相关[29, 30], 有研究显示, 肥胖者BMI指数越高, 有口臭的可能性越大, BMI可作为预测口臭的相关指数之一[31]。肥胖状态下口腔微生物的改变可能参与口臭的发生, 本研究从口腔微生物的角度为肥胖在口臭发病机制中的作用提供了新的线索。
本研究血链球菌的丰度在正常体重组唾液中较肥胖组高, 血链球菌是早期定植在口腔内的细菌之一, 是口腔内的常驻菌。血链球菌对大多数牙周可疑致病菌具有拮抗作用, 是一种牙周有益菌。流行病学调查显示肥胖是牙周病的一项危险因素[32], 正常体重者唾液中血链球菌的较肥胖者更多, 可能是其较低牙周病的患病风险的原因之一, 值得进一步研究。
综上所述, 本研究利用宏基因组学研究方法对肥胖者口腔唾液微生物的特点进行了探索, 研究样本量较小, 一定程度上揭示了肥胖者与正常体重者唾液微生物群落组成结构、基因及代谢通路的差异, 仍需扩大样本量进一步研究。此外, 同时留取受试者粪便样本以便研究口腔微生物群落与肠道微生物群落的关联性和交互影响以及在肥胖发病机制中的作用。对肥胖人群口腔微生物组成、基因及功能通路的初步探索为更深入研究口腔微生物群落与肥胖之间的相互关系提供参考, 为口腔微生物群落在疾病发生、发展及预警中的作用提供线索。
The authors have declared that no competing interests exist.
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|