

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
拓扑异构酶抑制剂通过ATM/ATR和NF-κB途径上调乳腺癌细胞MICA/B 的表达
[朱燕1 , 石永进2 , 赵玉亮1 , 朱平2, △  ]
]
]
|
|


乳腺癌是发病率最高的女性恶性肿瘤, 也是导致女性癌性死亡的最主要原因。患者免疫功能的强弱影响乳腺癌患者的生存和肿瘤进展[1], 机体消灭乳腺癌细胞的免疫反应主要依赖自然杀伤(natural killer, NK)细胞、CD8+ T细胞和γ δ T细胞[2, 3]。这三种细胞表面均表达抑制型受体和激活型受体, 当激活型受体表达增强时, 淋巴细胞清除体内肿瘤细胞的能力也增强。这些受体中较为重要的是NK细胞2族成员D(NK group 2 member D, NKG2D), 一种表达于NK细胞、CD8+ T细胞和γ δ T 细胞的激活型受体[4, 5]。NKG2D的配体为跨膜糖蛋白主要组织相容性复合体Ⅰ 类相关分子A和B(major histocompatibility complex class Ⅰ -related chain A and B, MICA/B)[5], 淋巴细胞通过NKG2D识别和杀伤表面表达MICA/B的肿瘤细胞。研究发现, MICA/B在一些肿瘤细胞表面表达的强弱不等, 表达MICA/B较强的肿瘤细胞容易被表达NKG2D的免疫效应细胞识别并杀伤。因此, 如果能够通过药物增强MICA/B在乳腺癌细胞的表达, 就能够促进免疫效应细胞对其的杀伤作用[6, 7, 8]。乳腺癌通常需要化疗, 一般认为化疗会破坏和降低机体的免疫功能, 但常用化疗药物多为DNA损伤诱导剂, 有研究显示一些DNA损伤诱导剂造成的细胞应激反应能够上调MICA/B表达[6, 7, 9], 如通过毛细血管扩张性共济失调突变和Rad3相关激酶(ataxia telangiectasia mutated and Rad3-related kinase, ATM/ATR)通路上调MICA/B表达[9]。那么, 这些化疗药物是否有可能促进肿瘤细胞的MICA/B表达, 从而诱导较强的机体免疫反应?
本研究检测6种常用乳腺癌化疗药物(足叶乙甙、拓扑替康、阿霉素、紫杉醇、氟尿嘧啶和顺铂)对乳腺癌细胞MICA/B表达的影响, 分析受到化疗损伤的乳腺癌细胞是否会改变免疫功能, 增强免疫系统对乳腺癌的识别, 并了解化疗药物通过哪些途径参与这一作用, 以期为临床化疗药物在直接杀灭乳腺癌细胞外, 还能诱导并增强机体的免疫反应提供一些启示。
人乳腺癌细胞系MCF-7和SK-BR-3购自中国医学科学院细胞资源中心。细胞系用含10%(体积分数)灭活胎牛血清(Gibco公司)的伊思柯夫改良培养液(Iscove’ s modified Dubecco’ s medium, IMDM)培养, 置于37 ℃含5%(体积分数)CO2的恒温孵箱中孵育。足叶乙甙、拓扑替康、阿霉素、紫杉醇和氟尿嘧啶购自南京凯基生物公司, 顺铂购自山东齐鲁制药公司, 咖啡因和吡咯烷二硫氨基甲酸酯(pyrrolidine dithiocarbamate, PDTC)购自Sigma-Aldrich 公司。
人乳腺癌细胞系MCF-7和SK-BR-3分别用足叶乙甙、拓扑替康、阿霉素、紫杉醇、氟尿嘧啶和顺铂在IMDM培养液培养。药物浓度分别为足叶乙甙1、5、20、100 μ mol/L, 拓扑替康0.5、2、10、25 μ mol/L, 阿霉素0.25、0.5、1、2 μ mol/L, 紫杉醇2、10、50、150 nmol/L, 氟尿嘧啶4、8、80、400 μ mol/L, 顺铂1、5、10、25 μ mol/L。
用Trizol提取细胞总RNA(2 μ g), 用Moloney鼠类白血病病毒逆转录酶和随机引物(Promega公司)进行逆转录。用SYBR Green和ROX荧光定量PCR试剂盒(Promega公司)在7300实时PCR系统(美国应用生物系统公司)进行PCR, 共40个循环:94 ℃ 25 s, 58 ℃ 25 s, 72 ℃ 32 s。cDNA重复三份扩增, PCR产物用测序验证(上海生工公司)。PCR引物为:MICA(158 bp):F-5' CCAGAGACTTGAC-AGGGAAC 3', R-5' CCCCATCGTAGTAGAAATGCT 3'; MICB(156 bp):F-5' CCGAGGACTTGACAGAGAA-TG 3', R-5'CCCCATCGTAGTAGAAATGCC 3'; GAPDH(180 bp):F-5' ATGGGGAAGGTGAAGGTCGG 3', R-5' GACGGTGCCATGGAATTTGC 3'。
定量RT-PCR以GAPDH作为内参照, MICA和MICB mRNA相对量用
不同药物处理乳腺癌(MCF-7、SK-BR-3)细胞后, 用2.5 g/L胰蛋白酶消化并收获, 磷酸盐缓冲液(phosphate buffer saline, PBS)冲洗。将细胞与标记藻红蛋白(phycoerythrin, PE)的MICA/B抗体(clone 6D4, BD公司)或IgG2a 同型抗体(isotype, BD公司), 在4 ℃避光孵育30 min, PBS冲洗后用FACS-Aria流式细胞仪(BD公司)检测。荧光强度计算:药物处理组细胞的荧光强度减掉同型对照抗体的荧光强度得到平均荧光强度(mean fluorescence intensity, MFI), 与未处理组细胞的平均荧光强度的比值为相对平均荧光强度。实验重复3次得到标准差。
抗体双染方法检测MCF-7细胞中存活和凋亡细胞表面MICA/B蛋白的表达:细胞用2.5 g/L不含EDTA的胰蛋白酶收获并用PBS冲洗。之后同时用Annexin V-FITC(Annexin V-FITC凋亡试剂盒, 南京凯基生物公司)和抗MICA/B抗体(或IgG2aB作为对照)双染色。FACSAria流式细胞仪检测。
乳腺癌细胞MCF-7先用不同浓度的咖啡因(1、5、10 mmol/L)处理2 h或PDTC(10、50、100 μ mol/L)处理1 h, 之后再与足叶乙甙(100 μ mol/L)孵育24 h, 收获细胞。分别用实时定量RT-PCR和流式细胞术检测各组细胞MICA/B mRNA和蛋白的表达, 比较加入咖啡因或PDTC前后MICA/B表达的变化。
乳腺癌MCF-7细胞培养液中加入足叶乙甙(终浓度100 μ mol/L), 分别收获孵育0.5、1、2 h的细胞进行凝胶阻滞实验(electrophoresis mobility shift assay, EMSA), 用NE-PER® 细胞核和胞浆提取试剂盒(赛默飞世尔科技公司)提取细胞核。蛋白质用Pierce® BCA蛋白分析试剂盒(赛默飞世尔科技公司)定量。用JASPAR数据库(http://jaspar.binf.ku.dk/)确定NF-κ B结合序列, 在MICA和MICB基因的第一个内含子中含有一段序列(5' ACATGGGGAAACCCCGCCTCTA 3')符合NF-κ B结合序列条件。采用第二代DIG Gel Shift Kit(罗氏公司)双链寡核苷酸末端标记地高辛-11-ddUTP作为探针, 核蛋白(12 μ g)与233 fmol地高辛标记的探针在含有1.0 μ L多聚左旋赖氨酸和1.0 μ L多聚[d(I-C)]的20 μ L反应液中孵育15 min。样本在聚丙烯酰胺胶上电泳2 h后转移至带正电荷尼龙膜上(Hybond-N+, Amersham公司), 用紫外交联仪进行交联。缓冲液冲洗后, 阻断液孵育30 min, 在阻断液中与碱性磷酸 酶标记的地高辛抗体(anti-digoxigenin-AP, 1:10 000)孵育45 min。冲洗后的膜用化学发光底物(chemiluminescence substrate, CSPD)均匀覆盖, 最后用Kodak Image Station 4 000 mm PRO检测发光信号。
采用SPSS 14.0软件完成统计分析, P< 0.05为差异有统计学意义。数据用均值± 标准差表示, 根据数据特征选择单样本t检验或单因素方差分析及两两组间比较方法完成假设检验, 两两比较采用LSD法。
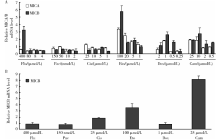
将乳腺癌常用化疗药物足叶乙甙、拓扑替康、阿霉素、紫杉醇、氟尿嘧啶和顺铂稀释为不同浓度, 孵育乳腺癌细胞系。结果发现, MCF-7在足叶乙甙、拓扑替康和阿霉素三种拓扑异构酶抑制剂多个浓度下, 均显著增加MICA和MICB mRNA的水平(图1A)。足叶乙甙在浓度为5、20、100 μ mol/L时, 与不加药组相比, MICA mRNA水平分别升高为(1.68± 0.17)、(2.54± 0.25)、(3.42± 0.15)倍(P< 0.05), MICB mRNA水平分别升高为(1.82± 0.24)、(1.56± 0.05)、(5.84± 0.57)倍(P< 0.05)。拓扑替康在浓度为10、25 μ mol/L时, MICA mRNA水平分别升高为(1.87± 0.49)和(1.94± 0.09)倍(P< 0.05); 在浓度为0.5、2、10、25 μ mol/L时, MICB mRNA水平分别升高为(1.54± 0.09)、(1.38± 0.48)、(2.77± 0.77)、(1.89± 0.08)倍(P< 0.05)。阿霉素在1 μ mol/L时, MICA mRNA水平升高为(1.84± 0.06)倍(P=0.002); 在浓度为0.5、1 μ mol/L时, MICB mRNA水平分别升高为(1.65± 0.15)和(2.62± 0.29)倍(P< 0.05)。其中, 足叶乙甙的作用最明显。氟尿嘧啶在相对高浓度(400 μ mol/L)时也显著上调MICB mRNA的表达, 顺铂在高浓度(25 μ mol/L)时轻度上调MICA mRNA的表达。
 | 图1 化疗药物对乳腺癌细胞MICA/B mRNA表达的作用Figure 1 Effects of chemotherapy agents on MICA/B mRNA expression in breast cancer cells Flu, fluorouracil; Pac, paclitaxel; Cis, cisplatin; Eto, etoposide; Dox, doxorubicin; Cam, camptothecin; MICA/B, major histocompatibility complex class Ⅰ -related chain A and B. A, MCF-7 breast cancer cells were incubated for 24 h with different chemotherapy agents; B, SK-BR-3 breast cancer cells were incubated for 24 h with different chemotherapy agents. |
SK-BR-3细胞系在加入足叶乙甙(终浓度100 μ mol/L)和拓扑替康(终浓度25 μ mol/L)后, MICB mRNA的表达水平显著升高(图1B), 与不加药组相比, 分别升高为(3.53± 0.45)倍(P=0.01)和(8.16± 0.4)倍(P=0.001)。顺铂能轻度上调其表达水平, 其他化疗药没有引起MICB mRNA表达升高。MICA mRNA在SK-BR-3不表达, 所有化疗药物均未能诱导MICA mRNA表达。
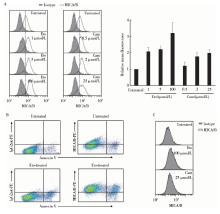
用流式细胞术检测拓扑异构酶Ⅱ 抑制剂足叶乙甙和拓扑替康是否影响MCF-7表面MICA/B蛋白的表达。足叶乙甙浓度为1、5、100 μ mol/L时, 与不加药组相比MICA/B蛋白的表达明显升高, 分别为(2.08± 0.26)、(2.24± 0.21)、(3.22± 0.62)倍(P< 0.05), 且呈现浓度依赖性。而拓扑替康处理后MICA/B 蛋白的表达仅轻度升高, 当药物浓度为2、25 μ mol/L时, 蛋白表达分别升高为(1.77± 0.25)、(1.98± 0.03)倍(P< 0.05)(图2A)。用Annexin V-FITC凋亡试剂盒和抗MICA/B抗体检测了足叶乙甙处理后的Annexin V阴性(存活)细胞和Annexin V阳性(凋亡)细胞的MICA/B蛋白水平, MICA/B蛋白的表达在存活细胞群和凋亡细胞群均升高(图2B)。SK-BR-3细胞经这两种化疗药处理后, 未检测到MICA/B蛋白(图2C)。
 | 图2 足叶乙甙和拓扑替康对乳腺癌细胞MICA/B表面蛋白表达的作用Figure 2 Changes of MICA/B surface protein expressions in breast cancer cells after etoposide or camptothecin treatment Eto, etoposide; Cam, camptothecin; MICA/B, major histocompatibility complex class Ⅰ -related chain A and B. A, MCF-7 cells were cultured for 24 h with etoposide or camptothecin before flow cytometric analysis of MICA/B surface expression. The right panel: Etoposide or camptothecin-induced MICA/B surface expression on MCF-7 was shown as relative mean fluorescence. B, Annexin V-FITC and anti-MICA/B mAb (or IgG2aκ ) staining of MCF-7 cells which were treated with etoposide (100 μ mol/L) or cultured in medium (control) for 24 h. C, SK-BR-3 cells were cultured for 24 h with etoposide or camptothecin prior to flow cytometric analysis of MICA/B surface expression. |
用不同浓度的咖啡因处理MCF-7细胞后再与足叶乙甙孵育, 结果发现足叶乙甙诱导的MICA/B mRNA和表面蛋白的表达均能被不同浓度的咖啡因抑制:当咖啡因浓度为1、5、10 mmol/L时, MICA mRNA相对表达水平由不用咖啡因处理组的 (3.75± 0.25)倍分别降为(0.89± 0.05)、(0.81± 0.02)、(0.48± 0.04)倍(P< 0.001), MICB mRNA由(6.85± 0.35)倍降为(1.36± 0.13)、(0.76± 0.06)、(0.56± 0.03)倍(P< 0.05)(图3A), MICA/B蛋白表达由(3.42± 0.05)倍降为(1.32± 0.03)、(1.21± 0.06)、(1.14± 0.03)倍(P< 0.001)(图3B), 提示MCF-7细胞的ATM/ATR通路对足叶乙甙诱导的乳腺癌细胞的MICA/B表达很重要。
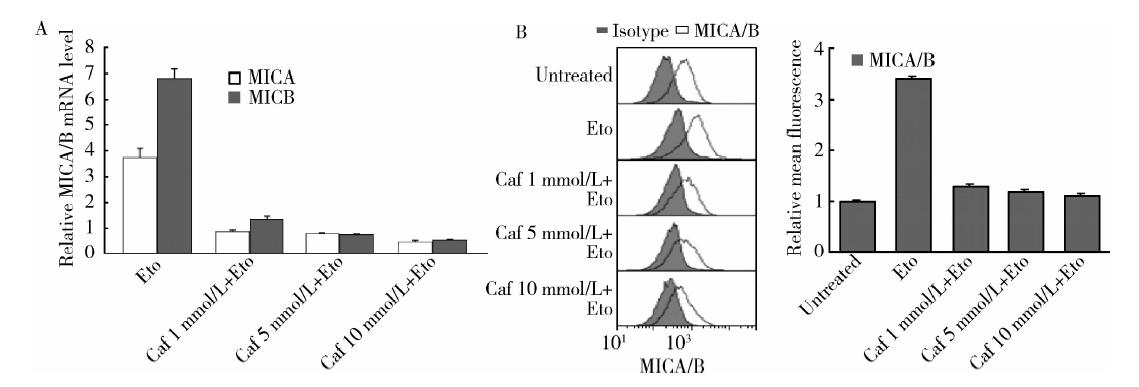 | 图3 ATM/ATR抑制剂咖啡因抑制了足叶乙甙对MCF-7细胞MICA/B的表达上调作用Figure 3 ATM/ATR inhibitor caffeine inhibited the upregulation of MICA/B expression by etoposide in MCF-7 cells Caf, caffeine; Eto, etoposide; MICA/B, major histocompatibility complex class Ⅰ -related chain A and B. A, MICA/B mRNA expression by real-time PCR; B, MICA/B surface protein expression by FACS. The right panel: MICA/B surface expression changes on MCF-7 were shown as relative mean fluorescence. |
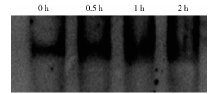
用不同浓度的NF-κ B抑制剂PDTC处理MCF-7细胞后再与足叶乙甙孵育。结果发现足叶乙甙诱导的MICA/B mRNA和表面蛋白的表达均能被不同浓度的PDTC抑制:当PDTC浓度为10、50、100 μ mol/L 时, MICA mRNA的水平由不加PDTC组的(4.46± 0.97)倍分别降为(2.8± 0.25)、(2.1± 0.02)、(1.72± 0.08)倍(P< 0.05), MICB mRNA由(8.11± 0.17)倍降为(4.58± 0.05)、(3.06± 0.51)、(1.02± 0.25)倍(P< 0.001)(图4A), MICA/B蛋白表达由(2.27± 0.12)倍降为(1.74± 0.05)、(1.33± 0.15)、(0.93± 0.05)倍(P< 0.001)(图4B), 提示NF-κ B参与这一过程。EMSA方法显示MCF-7细胞用足叶乙甙处理后, NF-κ B与MICA/B基因启动子区的结合活性升高(图5), 进一步提示NF-κ B参与了足叶乙甙对MICA/B表达的诱导作用。
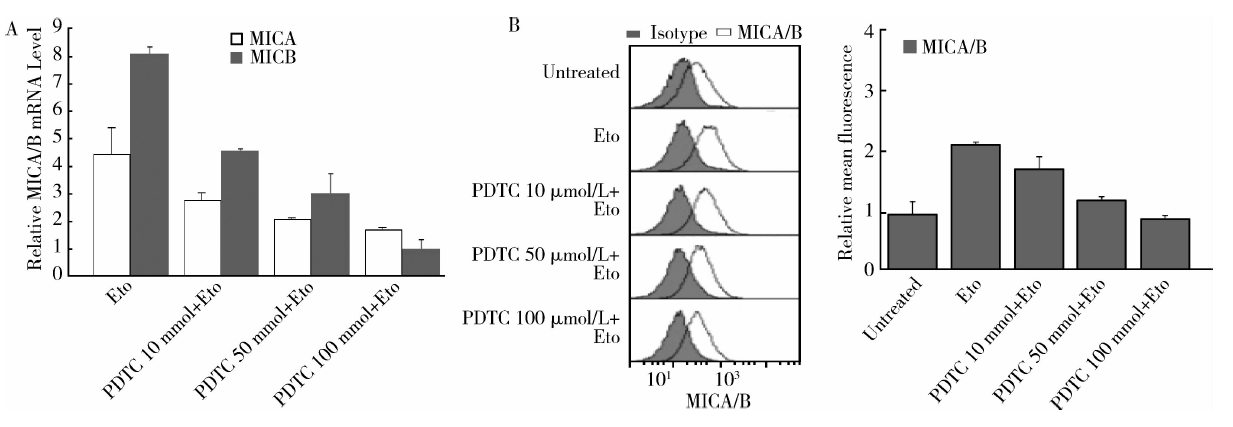 | 图4 NF-κ B抑制剂PDTC抑制了足叶乙甙对MCF-7细胞MICA/B的表达上调作用Figure 4 NF-κ B inhibitor PDTC inhibited the upregulation of MICA/B expression by etoposide in MCF-7 cells PDTC, pynolidine dithiocarbamate; Eto, etoposide; MICA/B, major histocompatibility complex class Ⅰ -related chain A and B. A, MICA/B mRNA expression by real-time PCR; B, MICA/B surface protein expression by FACS. The right panel: MICA/B surface expression changes on MCF-7 were shown as relative mean fluorescence. |
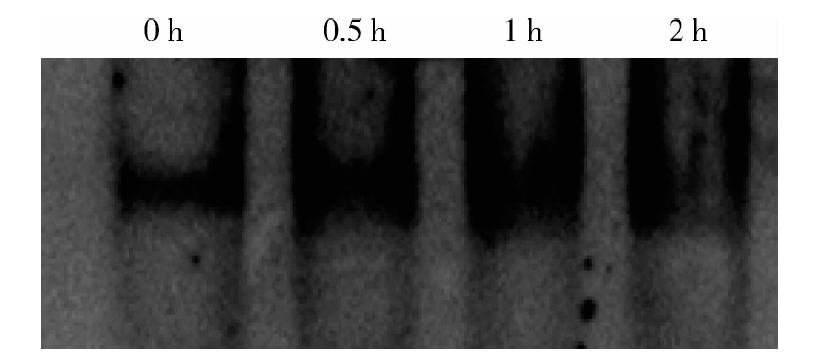 | 图5 足叶乙甙增强了NF-κ B与MICA/B基因启动子区的结合Figure 5 Etoposide enhanced the binding of NF-κ B to MICA/B promoter |
细胞表面的MICA/B是应激诱导基因, 其在肿瘤表面表达的强度直接与免疫系统对肿瘤细胞反应的强度有关。各种具有基因毒性的刺激物都有可能上调其在正常细胞和癌细胞的表达[9]。有研究提示, 某些特定试剂在某种癌细胞能够上调MICA/B表达, 例如:吉西他滨可增强非小细胞肺癌细胞MICA/B的表达[10], 组蛋白去乙酰化酶抑制剂可上调骨髓瘤和骨肉瘤细胞表面MICA/B的表达[6, 7]。阿非迪霉素或顺铂不能改变MICA/B在小鼠白血病细胞的表面蛋白水平, 博来霉素则轻度上调其表达[11]。临床上, 乳腺癌的治疗除了手术外, 经常需要长期化疗。通常认为化疗会破坏和降低机体的免疫功能, 但不化疗又往往难以抑制肿瘤生长, 究竟适度化疗是否会严重影响机体免疫反应, 还是有可能增强免疫功能是令人困惑的问题。常用化疗药物多为DNA损伤诱导剂, 它们是否也有可能促进乳腺癌的MICA/B表达, 反而能够诱导较强的机体免疫反应?本研究发现, 三种拓扑异构酶抑制剂足叶乙甙、拓扑替康和阿霉素显著上调MCF-7乳腺癌细胞MICA/B mRNA的表达, 同时也升高了细胞表面MICA/B蛋白表达水平, 而且存活癌细胞的MIC水平也升高。从临床治疗的角度来看, 很可能化疗药物在患者机体内除了杀伤乳腺癌细胞外, 还能够诱发乳腺癌细胞表面出现更丰富的MICA/B蛋白, 增强免疫细胞对化疗后残留肿瘤细胞的识别和杀伤。本实验中拓扑异构酶抑制剂的这种作用更为强烈, 存活和凋亡细胞MICA/B蛋白的表达均增高, 但另一个乳腺癌细胞系SK-BR-3则没有检测到MICA/B蛋白表达, 仅有MICB mRNA表达增高。这种蛋白和mRNA表达的不一致性在其他组织类型细胞也有报道, 可能还有转录后机制参与调控[12]。有报道热休克[13]、氧化应激[14]、炎症细胞因子[15, 16]、毒性药物[9, 11, 17, 18]引起的DNA损伤反应加强肿瘤细胞MICA/B的表达可能存在多种途径。其中, 一些研究显示, ATM/ATR引发的DNA损伤反应参与了化疗药物诱导的MICA/B表达[9, 11, 17, 18]。ATM和ATR属于磷脂酰肌醇3激酶相关蛋白激酶(phosphatidylinositol 3-kinase-related kinase, PIKK)家族, 是DNA损伤检查点的主要成员, 它们可以被不同类型的DNA损伤所激活, 通过磷酸化相应的下游蛋白Chk1和Chk2等, 调节细胞周期各个检查点, 引起细胞周期阻滞, 使DNA损伤得以修复。ATM和ATR在维持基因组的稳定性中起到至关重要的作用, 有研究显示, 在人纤维母细胞和小鼠肿瘤细胞系, NKG2D配体的表达依赖于DNA损伤反应途径中的ATM/ATR信号, 其表达能够被ATM/ATR抑制剂咖啡因所阻止[5]。本研究分析了ATM/ATR抑制剂咖啡因对拓扑异构酶抑制剂诱导癌细胞MICA/B高表达的影响, 结果表明ATM/ATR抑制剂咖啡因可以阻断拓扑异构酶抑制剂诱导的MICA/B mRNA和蛋白表达。拓扑异构酶抑制剂能强有力地诱导双链DNA断裂, 而ATM的激活需要双链DNA断裂的存在[19], 这可能是多种拓扑异构酶抑制剂均能升高MICA/B mRNA表达的原因。
为阐明拓扑异构酶抑制剂为何可以使乳腺癌细胞MICA/B表达增高, 我们还分析了NF-κ B与MICA/B基因表达的关系。NF-κ B转录因子家族是许多信号传递途径中的重要交集点, 它调控的基因对于初始和适应性免疫反应、细胞迁移和凋亡有着重要作用[20]。Lin等[16]认为肿瘤坏死因子a在内皮细胞通过NF-κ B上调MICA的表达, Schilling等[21]认为NF-κ B受抑会降低MICA在癌细胞的表达, 这些均提示化疗药物有可能通过NF-κ B上调MICA/B表达。我们发现NF-κ B抑制剂PDTC能抑制足叶乙甙对MICA/B的表达诱导作用, 提示药物可能通过转录因子NF-κ B上调MICA/B的表达。有研究显示, 在佛波酯(phorbol-12-myristate-13-acetate, PMA)激活的T细胞中, NF-κ B可以通过与MICA内含子1上的特定序列结合来调节MICA表达[22]。我们的凝胶阻滞实验提示, 足叶乙甙处理后, NF-κ B与MICA/B基因启动子区的结合活性明显升高。通过ATM/ATR途径和NF-κ B途径都可以增强乳腺癌细胞MICA/B的表达, 但这两种途径间是否存在联系?Cerboni等[23]的研究显示, MICA激活T细胞的表达是通过ATM-NF-κ B途径介导的。但乳腺癌细胞是否经过同样途径尚需研究, 我们将在后续的研究中证实EMSA实验中所检测的这段启动子序列的活性, 以说明NF-κ B介导了乳腺癌细胞MICA/B表达的过程。
总之, 拓扑异构酶抑制剂可以明显增强乳腺癌细胞MICA/B表达, 患者使用这种化疗药物后, 机体免疫系统可能能够更好地识别和杀灭乳腺癌细胞, 改善治疗效果, 这一MICA/B表达增强的作用可能是通过ATM/ATR和NF-κ B途径实现的。这些发现可以为临床上将化疗和免疫治疗相结合治疗乳腺癌提供参考。
The authors have declared that no competing interests exist.
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|