
{kind=link}
{kind=link}
基因诊断Caroli综合征合并常染色体隐性遗传性多囊肾1例及文献回顾
[杨夕樱1, △ , 朱灵平2 , 刘雪芹1, △  , 张春雨
, 张春雨1 , 姚勇1 , 吴晔1 ]
, 张春雨|
|


* 现在中国人民解放军总医院儿科
报道1例基因学诊断明确的Caroli综合征合并常染色体隐性遗传性多囊肾(autosomal recessive polycystic kidney disease, ARPKD)中国婴儿病例。本例患儿为8个月男婴,隐匿起病,以无症状性肝、肾肿大为主要临床表现,影像学检查提示肝内胆管囊状扩张、双肾多囊性改变,血生化检查示肝、肾功能正常,肝脏纤维化4项标记物血清水平均升高,肾早期损伤指标阳性,基因检测证实为多囊肾/多囊肝病变1基因(polycystic kidney and hepatic disease 1, PKHD1)错义杂合突变c.9292G>A、c.2507T>C,分别来自于患儿父母。前者为新发突变,经预测软件验证存在高致病性,后者目前见于1例中国双胞胎报道,可能为中国人群独有,基因型与临床表型之间关系未明。患儿出院后11个月随访,肝、肾功能正常,尚无严重并发症发生。婴儿期起病的Caroli综合征合并ARPKD临床表现隐匿,肝、肾肿大通常为唯一表现。通过对该患儿的临床表现系统性回顾分析以及文献复习可以得出,Caroli综合征合并ARPKD的肝、肾影像学表现具有特征性,在影像基础上联合基因检测手段有利于明确诊断并与其他肝、肾囊性纤维化疾病进行鉴别。针对此类患儿的长期管理,应包括定期随访,密切监测肝、肾功能及各种并发症的发生,适当干预,同时做好患儿家庭的遗传咨询工作。
This case report is about one genetically specified diagnosed infant case of Caroli syndrome with autosomal recessive polycystic kidney disease (ARPKD) in China. The patient in this case report was an eight-month infant boy with an atypical onset and the main clinical manifestation was non-symptomatic enlargement of the liver and kidneys. The imaging study demonstrated a diffused cystic dilatation of intrahepatic bile ducts as well as polycystic changes in bilateral kidneys. The basic blood biochemical tests indicated a normal hepatorenal function. Four serum biomarkers of hepatic fibrosis were all elevated and the urine test for an early detection of the renal injury was positive. The genetic sequencing proved two heterozygous missense mutations of polycystic kidney and hepatic disease 1 ( PKHD1) gene, c.9292G>A and c.2507T>C, inherited from each of his parents respectively. The former was a novel mutation that had been verified as disease causing through the predicting software while the latter had been reported from one recent case study on Chinese twins, which was possibly unique among Chinese population. The relations between the gene type and the clinical phenotype were not clarified yet. Up till a follow-up eleven months later after the discharge, the patient had a normal hepatorenal function without occurrence of any severe complication yet. The clinical symptoms of Caroli syndrome with ARPKD at infant stage were atypical and the enlargement of liver and kidney was usually the sole symptom. From the above systematic retrospective clinical analysis, as well as the relevant literature review, it’s been concluded that the features of the hepatorenal images in patients with Caroli syndrome and ARPKD were distinctive. Genetic testing combined with the imaging study benefits a definite diagnosis as well as a diffe-rentiation from other hepatorenal fibrocystic diseases. Specific to the long-term management of this kind of patients, it’s necessary to schedule a regular follow-up to monitor the hepatorenal function and the occurrence of various complications for an appropriate intervention, meantime to devote efforts to the genetic counseling work for the patients’ family.
先天性肝内胆管囊状扩张症又称Caroli病, 合并肝纤维化则称为Caroli综合征。Caroli综合征为常染色体隐性遗传, 由多囊肾/多囊肝病变1基因(polycystic kidney and hepatic disease 1, PKHD1)突变所致, 该基因同时也是常染色体隐性遗传性多囊肾(autosomal recessive polycystic kidney disease, ARPKD)的致病基因[1]。根据北美流行统计学数据, ARPKD在活产新生儿中发病率约为1/20 000[2], 其中合并Caroli综合征者可高达70%以上[3, 4]。关于本病的临床及基因突变特点, 国外已有较多研究[3, 5], 但迄今国内明确基因诊断的病例尚少。本文报道1例8个月男婴, 隐匿起病, 以无症状性肝、肾肿大为主要临床表现, 影像学表现典型, 基因检测证实为PKHD1基因复合杂合突变。本研究结合此病例, 简述Caroli综合征合并ARPKD的临床特点、分子生物学机制及诊疗方面的新进展, 以提高对本病的认识。
患儿男, 8个月, 因院外体检发现肝、肾肿大入院。既往无肝、肾疾病史。患儿为G1P1, 足月因“ 羊水偏少” 剖宫产, 出生体重2.55 kg, 母亲孕期轻度贫血, 未予治疗, 孕期胎儿超声未见异常。患儿体格及智力运动发育同正常同龄儿, 家族成员无肝、肾疾病史, 父母腹部超声正常。
体格检查:体重9 kg, 身长73 cm, 血压90/60 mmHg(1 mmHg=0.133 kPa), 精神反应好, 皮肤略苍白, 巩膜、皮肤无黄染, 心、肺无异常, 腹稍膨隆, 肝肋下约3 cm, 脾肋下约1 cm, 质软、边钝, 双侧下腹部各可触及1个直径约3 cm类圆形包块, 质地中等、表面光滑、活动度差、无压痛, 腹部移动性浊音阴性, 全身无水肿, 神经系统无异常。
血常规:白细胞计数12.8× 109/L, 血红蛋白70 g/L, 平均红细胞体积 54.9 fL, 血小板计数314× 109/L。尿常规:白细胞 10~12/HP(高倍视野)。便常规及潜血(-)。
血生化检查:谷丙转氨酶20 IU/L, 谷草转氨酶37 IU/L, 白蛋白43 g/L, 碱性磷酸酶166 IU/L, 谷氨酰转肽酶13 IU/L, 总胆红素7.9 μ mol/L, 直接胆红素0.2 μ mol/L, 总胆汁酸18.8 μ mol/L, 血肌酐28.1 μ mol/L, 尿酸434 μ mol/L, 尿素氮4.2 mmol/L, 胱抑素C 1.11 mg/L(正常值0.59~1.03 mg/L), 碳酸氢根 16.8 mmol/L, 血电解质正常, β 羟丁酸 1.56 mmol/L, 乳酸4.52 mmol/L, 总铁结合力90.3 μ mol/L, 铁蛋白3.8 μ g/L, 血清铁3.4 μ mol/L。
肝纤维化4项:Ⅲ 型前胶原N端肽53.94 μ g/L(正常值0~30 μ g/L), 血清透明质酸165.57 μ g/L(正常值0~100 μ g/L), 层粘连蛋白58.37 μ g/L(正常值0~50 μ g/L), Ⅳ 型胶原60.48 μ g/L(正常值0~30 μ g/L)。
肾早期损伤指标:尿微量白蛋白41.80 mg/L(正常值0~19 mg/L)。血及尿遗传代谢病筛查未见异常, 肿瘤标志物均(-)。骨髓穿刺检查提示缺铁性贫血。
心电图示窦性心动过速, 超声心动图提示左心室轻度扩大, X线胸片未见异常。
腹部超声:左肾11.2 cm× 4.8 cm, 右肾9.7 cm× 4.3 cm, 双肾弥漫性增大, 实质回声不均, 正常皮髓质结构消失, 双肾实质内可见多发细小囊腔, 右侧较大者约0.3 cm× 0.2 cm, 左侧较大者约0.2 cm× 0.2 cm, 双肾髓质区呈弥漫性密集的强回声光点, 后见彗星尾征, 边缘可见一圈肾皮质回声, 肾盂、肾盏无扩张, 双侧未见扩张的输尿管。肝肋下2.3 cm, 剑突下3 cm, 肝实质回声异常粗糙, 呈网格样, 肝内胆管呈囊样扩张, 较大者约1.3 cm× 0.7 cm× 0.8 cm, 肝门区及胆总管未见扩张, 胆囊透声可, 脾肋下1 cm, 实质回声均匀。
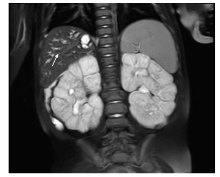
磁共振胰胆道造影(magnetic resonance cholangiopancreatography, MRCP):肝体积增大, 肝内胆管轻度扩张, 胆管走行区周围多发囊状、片状呈长T1长T2信号, 较大者0.8 cm, 呈分支状, 增强扫描未见明显强化; 胆囊、胆总管、胰腺、脾未见明显异常。双肾体积增大, 边缘欠光滑, 实质内呈fsT2WI高信号, T1WI低信号, 皮质较薄, 最厚处约0.3 cm, 髓质增厚, 增强扫描呈稍低强化(图1)。
 | 图1 磁共振压脂T2WI序列提示多发肝内胆管囊、片状扩张(白色箭头处)及双肾多囊性改变Figure 1 MRI T2WISTIR image demonstrates sacular and fusiform dilatation of multifocal intrahepatic bile ducts (white arrow) and the polycystic changes in both kidneys |
分别采取患儿及其父母的外周血标本提取DNA, 应用第二代测序方法对囊性肾脏病PKHD、PKD基因进行全外显子测序并使用Sanger法测序验证。结果显示, PKHD基因外显子区域发现两处杂合突变点:c.9292G> A(鸟嘌呤> 腺嘌呤)来自其母, 导致了p.G3098R(甘氨酸> 精氨酸)氨基酸改变; c.2507T> C(胸腺嘧啶> 胞嘧啶)来自其父, 导致了p.V836A(缬氨酸> 丙氨酸)的氨基酸改变(图2)。
 | 图2 患儿及其父母PKHD1基因测序图Figure 2 gene sequencing of the patient and his parents A and B show the c.9292G> A mutation of the patient and his mother, respectively; C and D show the c.2507T> C mutation of the patient and his father, respectively. Reference sequences are complementary sequences. |
入院后给予抗感染、铁剂及利胆药物, 复查尿常规正常, 住院治疗2周出院, 出院诊断:Caroli综合征合并ARPKD, 慢性肾脏病, 尿路感染, 缺铁性贫血。出院后11个月随访, 患儿体重增长, 血压正常, 血红蛋白117 g/L, 复查肝、肾功能, 胱抑素C, 总胆汁酸均正常, 腹部超声检查结果与病初大致相同。
本例8个月龄男婴以无症状性肝、肾肿大为临床表现, 腹部影像学提示肝内胆管囊状扩张, 肝实质弥漫性病变, 双肾弥漫性增大, 皮髓质分界不清伴有多囊性改变, 实验室检查结果提示肝纤维化, 基因检测为PKHD1基因复合杂合突变, 因此Caroli综合征合并ARPKD的诊断明确。本例患儿婴儿期发病, 无家族史, 肝、肾同时受累, 根据影像学及基因检测结果, 可除外常染色体显性遗传多囊肾、肾单位肾痨以及其他肾小球囊性肾疾病等肝肾囊性纤维化疾病(hepatorenal fibrocystic diseases, HRFCD)。
Caroli综合征及ARPKD常同时发生, Gunay-Aygun等[5]对一组平均年龄12.7岁的73例患者的研究中, 发现二者的并发率高达71%。本病中的肝受累以肝脏增大、中型胆道非梗阻性囊样扩张及先天性肝纤维化为特征, 组织病理学特征为胆管板发育异常[1]。肝病变的临床表现较隐匿, 多无肝酶升高及合成功能下降导致的低白蛋白血症及凝血因子减少, 而常见逆行性胆管炎及门脉高压, 门脉高压并发症所致消化道出血是导致死亡的危险因素[6, 7]。ARPKD的肾脏病变为肾髓质集合管囊性变, 重者皮髓质均受累, 最终导致高血压及慢性肾功能不全, 胎儿期发病最为常见, 可伴肺发育不良, 约30%~40%于围生期死亡[6], 存活者约25%至11岁前发展为终末期肾脏病, 需进行肾移植, 而新生儿期之后发病者存活至32岁时也有25%需要肾替代治疗[8]。临床研究多认为, 本病中的肝脏与肾脏病变无相关性, 二者独立发生及进展, 肾衰竭早于肝衰竭[5, 6]。Gunay-Aygun等[5]的研究显示, 73例患者中10例进行了肾移植, 1例接受肝、肾联合移植。本病中的肾移植预后与其他原发肝、肾疾病移植的疗效相当, 有报道本病肾移植及肝移植后的5年死亡率分别为6%及20%[9]。
本病的临床诊断主要依靠B超等影像学检查, 肝脏病变特征为肝内胆管囊样扩张, 存在肝纤维化及门脉高压时可有相应影像学表现; 肾脏病变特征为双侧肾回声增强伴皮髓质分界不清。高分辨超声、计算机断层扫描以及磁共振等影像技术可提高诊断的敏感性, 尤其磁共振胆道造影可清晰显示肝内胆管扩张情况。本病的临床表现隐匿, 可无任何临床症状, 婴儿期常以发现肝、肾肿大为唯一临床表现[6], 本例患儿亦具有此特点, 最终经影像学检查及基因诊断确诊。本例患儿肝脏病变突出, 影像学检查示肝内胆管扩张, 肝实质回声粗糙并呈网格样, 肝纤维化4项血清学指标均升高提示存在肝纤维化, 超声发现脾肿大, 提示已存在门脉高压, 外周血血小板及白细胞均正常, 提示尚未发生脾功能亢进。患儿尿中曾见较大量白细胞, 提示泌尿系感染可能。肾脏超声见双肾髓质区呈弥漫性密集的强回声光点伴彗星尾征, 提示存在肾钙化。入院后患儿血压正常偏高, 肌酐及尿素氮均在正常范围, 但微量白蛋白尿、代谢性酸中毒及高尿酸血症仍提示肾损伤, 因未能成功留取24 h尿量, 未能进行肌酐清除率检测。
Caroli综合征及ARPKD均是由于PKHD1基因突变导致的一种早发性HRFCD, 该基因位于第6号染色体上, 位置为6P21.1-P12。致病基因PKHD1是由469 kb碱基对构成的包含66个外显子的较大基因, 其编码的纤囊素(fibocystin/polyductin)蛋白由4 074个氨基酸组成, 是在肾小管和胆管上皮细胞表达的膜蛋白, 定位于初级纤毛, 该蛋白突变影响肾小管和胆管上皮的黏附、排斥及增殖[10], 其确切作用机制尚在研究中。本病为常染色体隐性遗传, 目前PKHD1基因已报道突变300余种, 主要为错义突变, 少数导致蛋白截断突变[3, 11], 本例患儿发现的c.2507T> C及c.9292G> A复合杂合突变均为错义突变, 两个突变分别来自患儿父母, 分别导致第24个外显子上第836个密码子编码的缬氨酸变为丙氨酸, 第58个外显子上第3 098个密码子编码的甘氨酸变为精氨酸。c.2507T> C突变既往报道仅见于我国山东一对异卵双胎病例[12], 其他种族未见报道, 提示其可能为中国人特有突变, 而c.9292G> A文献未见报道, 经PolyPhen2、SIFT及Mutation taster预测软件验证具有高致病性。
本病基因型与临床表型的关系国外已有大量研究, 发现二者无明显相关性, 但导致纤囊素蛋白截断的纯合突变病情较重, 常于围生期死亡[13, 14]。近年报道的我国山东2例10岁异卵双胎男孩, 为c.2507T> C错义及c.2341C> T无义复合杂合突变, 其中弟弟于1岁时发现无症状脾肿大, 5岁时出现呕吐及腹部肿物, 确诊为Caroli病, 2年后发生肝硬化、脾功能亢进及多囊肾改变, 而哥哥仅有肝内胆管扩张, 至10岁基因诊断明确时仍未出现临床症状[12], 也提示基因型与表型间缺少相关性。本例患儿虽与上述2例有部分相同突变, 与之不同的是, 本例患儿表现为婴儿期肝、肾同时受累, 再次显示本病基因型与表型关系的复杂性, 推测可能有其他基因参与了PKHD1基因的表达调控。迄今国内进行基因诊断的病例较少, 国人PKHD1基因突变类型与表型的相关性有待进一步研究。文献报道本病基因突变的检出率仅为80%左右[14, 15], 因此欧美专家共识指出, 现阶段ARPKD的确诊主要还是依靠影像学检查, 基因诊断作为重要补充[16], 也有学者提出将PKHD1基因突变检测、腹部超声检查及磁共振检查三者结合, 作为早期诊断ARPKD的有效手段[17], 同时, 基因诊断在本病与其他HRFCD的鉴别中也起到至关重要的作用。
本例Caroli综合征伴ARPKD病例, 肝、肾双器官病变典型, 鉴于本病肝、肾病变独立发生发展, 因此一旦确诊应长期密切随访。建议针对肝脏病变, 应每年检测血常规及血小板, 超声监测肝内外胆管扩张情况及脾脏大小。本例患儿目前已有肝纤维化4项实验室指标阳性, 脾脏稍大, 提示已存在门脉高压, 随访中应注意监测上述指标变化, 准确把握门脉高压的发生发展及外科治疗, 注意食道、胃底静脉曲张情况, 预防消化道出血, 及时发现和治疗复发性逆行性胆管炎。肾脏方面, 随年龄增长可出现高血压、肾功能进行性减退, 可发生泌尿系感染, 随访中应注意监测血压及肾功能情况, 积极监控感染, 根据病情适时进行肝、肾替代治疗。本患儿确诊后近1年, 病情尚稳定, 仍在密切随访中。
婴儿期起病的Caroli综合征合并ARPKD症状隐匿, 临床医生要提高对本病的认识, 根据肝、肾影像学特点进行初步诊断, 基因检测可确诊并除外其他HRFCD。对Caroli综合征合并ARPKD的患儿应定期随访, 密切监测肝、肾功能及各种并发症的发生, 适当干预, 同时做好患儿家庭的遗传咨询工作。
The authors have declared that no competing interests exist.
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|