{kind=link}
一种基于图塌缩的药物分子检索方法
[瞿经纬1 , 吕肖庆1, 2, △  , 刘振明
, 刘振明3 , 廖媛1 , 孙鹏晖1 , 王蓓1 , 汤帜1, 2 ]
, 刘振明|
|


医药信息检索旨在服务于大型的医药数据库、信息库和知识库, 需要融合药学、信息检索和机器学习等跨领域的先进技术, 基于化学分子结构式的检索方法已成为医药信息检索的关键技术之一, 现有检索系统多采用间接方式简化对分子结构的描述, 其检索结果的准确度也相应地受到了影响。图相似性的比较可以充分利用分子的空间结构信息, 实现准确、高效的检索, 但图计算的复杂度一直阻碍着该方法的应用。图形识别技术可看作是基于内容的图像检索的一个分支。
针对化学分子结构式识别与检索的研究可被大致分成三类:基于序列的方法、基于分子指纹(molecular fingerprint)的方法和基于图的方法。许多生物和化学信息用连续字符串来表示分子, 如最具代表性的SMILES(Simplified Molecular Input Line Entry System)[1]语言, 但此类编码方法忽略过多的结构信息。分子指纹[2]是另一类简单的分子编码方式, 如拓扑指纹(topological fingerprints)[3]将代表分子特征的键径转换为给定数量的连接键, 但分子指纹在精确描述空间结构方面仍有一些困难。图是在化学信息学和生物信息学领域中被广泛应用的一种通用的数据结构[4], 但当分子结构式随着原子数增加变成大图后, 该方法会因计算代价的瓶颈而无法取得很好的实用性。为此, 研究人员一直在探索大图的简化方法或更加有效的特征转换方法。
基于图的方法可以分成四个子类, 即图描述符、基于相似性的图挖掘、图嵌入(graph embedding)以及图核方法。图描述符涉及的范围较广, 包括从一维的统计[5]到二维的拓扑指数[6], 再到复杂的三维描述[7]。图挖掘旨在研究相似的化合物或者预测分子的物化和生物性质, 所应用的常规技术包括最大公共子图[8]、频繁子图挖掘[9]和编辑距离[10]。图嵌入是指使用向量来表示图。为了将图与机器学习方法相结合, 需要将每一个图映射到一个显式的向量空间[11]。在新的向量空间中, 被嵌入图通过所获编码进行差异比较[12], 从而度量出属性的统计特征。在图嵌入方法中会因将图“ 压缩” 成简洁的向量表示而丢失部分结构信息, 为了避免这一严重缺陷, 图的核方法被提出。该类方法采用了更加完备的数学框架, 如把图之间的相似性测量定义为希尔伯特空间(Hilbert space)的点积运算。Gaü zè re等[13]采用了由严格子树集定义的模式袋(bag of pattern), 即所有具有最多六个节点的标签树的集合。环信息在分子性质中具有特别的影响力。环模式核(cyclic pattern kernel)[14]把一个图分解成一个具有环结构的集合和一个与不在环中的原子和边相对应的集合。
尽管研究人员已经对化学分子结构式进行了数十年的研究, 但仍存在诸多挑战, 比如在对大分子进行搜索时难以承受的计算代价, 以及缺少一种对结构分类的标准等。总体上看, 现有图匹配和图挖掘方法仅限于小分子结构, 难以处理大型分子数据集中的复杂结构, 此外, 虽然已有个别技术被用于图压缩、简化分子的结构信息, 但大多局限于有限的且是预定义的结构模板, 尚不能高效处理种类繁多的子结构, 特别是非环结构。据此, 本研究对分子结构式的多样性和内在特性进行了深入的分析, 提出一种子图塌缩的方法, 该方法可有效简化大图描述, 能够根据结构式的特点并结合子图匹配与频繁子图挖掘等方法对大图进行多级塌缩, 使得分子的匹配计算在多维度信息支持下变得可行, 进而有效地提高了分子识别与检索的准确率。
本文针对药物分子结构式的特点, 建立了一个完整的药物分子检索系统, 其框架如图1所示, 主要模块包括:多种模态的分子结构式查询输入(文本、图片、手绘图)、底层图形信息检测与中层图形特征提取及描述、图空间结构与拓扑关系分析、基于图密度与子图占优性分析的多级塌缩、超图构造、图匹配与相似性度量。
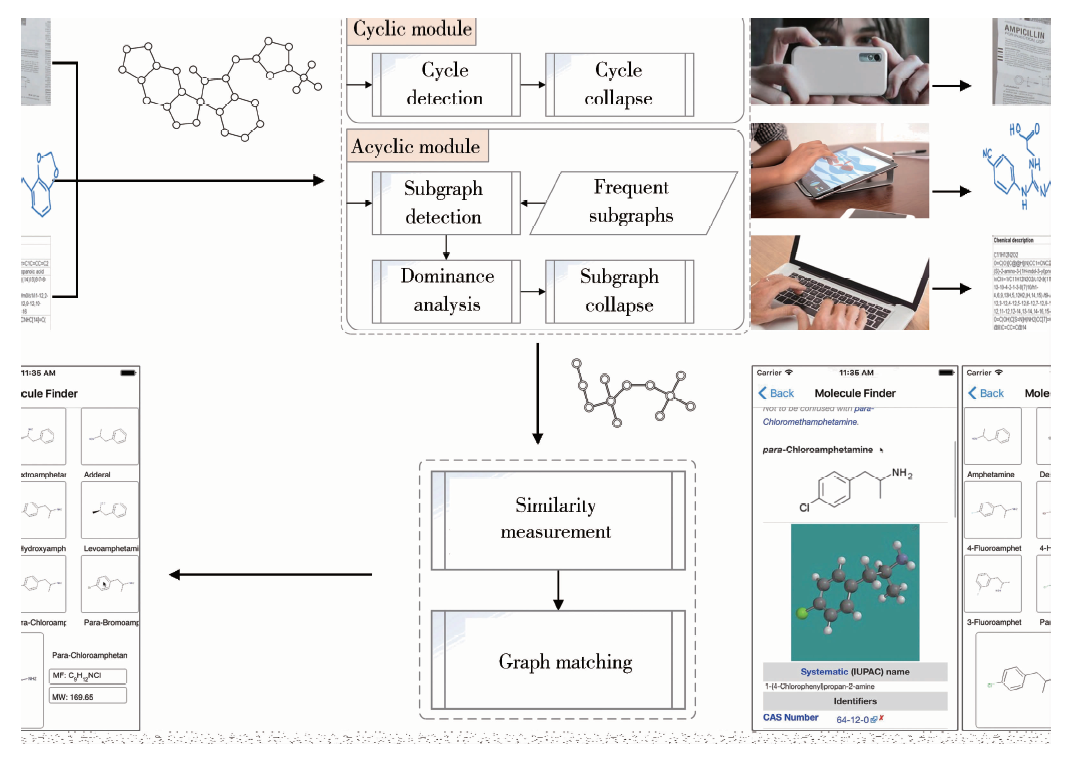 | 图1 基于图相似度的医药信息检索系统示意图Figure 1 Workflow of CSF retrieval system |
在本文方法中, 化学分子结构式可通过多种不同方式输入至检索系统。对于利用智能手机拍照的方式, 本文采用了一种规则化的方法从文本、图片和手绘图中提取分子结构式, 包括图像预处理、边缘检测、符号识别、结构式重建等关键技术。对于通过平板电脑手绘的方式, 本文针对性地采用了一些技术, 例如根据对时间序列的分析检测拐点、结合交互手势区别符号和线段、利用先验化学知识重建布局等。
结构式可表示为无向属性图, 其中顶点表示原子, 顶点间的边表示原子间的键。
定义1: 一个化学分子结构式定义为一个无向属性简单图
采用无向图来表示结构式是一种沿用多年的常规表达方式, 但其所得到的图结构会面临一个重要的挑战, 即在原子数量增多后, 相应图的计算复杂度会呈指数级增加, 因此, 为提升检索效率, 本文针对结构式的特点探索大图简化的途径, 根据一个分子是否包含环状结构将其分为有环分子和无环分子两类, 分别进行处理。
环状结构是结构式中的重要常见结构, 其具有相对稳定的化学性质, 采用有效方式对其进行塌缩可以简化分子的描述。这里的环状结构是指:每个顶点分别通过一条边和两个其他顶点相连, 即每个顶点的度数为2。要采用塌缩算法必须实现准确的环结构检测方法:对于任意给定的一个分子图结构, 使用RDKit[15]方法提取其中的环结构; 将每个环塌缩为一个超级顶点, 并将其特征赋予到顶点属性中。同时, 为了准确描述该图中多个环结构之间的拓扑关系(如常见的相离、相切、相交、多重相交等空间位置关系), 本文构建了对应的环图
不同于有环分子具有典型的环结构, 无环分子的子结构富有多样性, 为此需要借助更高层面的统计规律。例如, 对于给定的图数据集, 利用频繁子图挖掘技术检测具有代表性的非环结构, 由于候选非环子图之间存在多重选择性, 需要对其交叠情况进行更加细致的分析。为便于后文对上述方法进行详细介绍, 在此给出频繁子图的定义。
定义2:将一个频繁子图表示为具有支持度H(Gf)的图
1.3.1 基于频繁子图挖掘的非环结构筛选 与环状结构稳定的布局特征不同, 大多数无环分子中的子结构不仅数量众多, 而且结构各异, 相互之间的空间布局关系也远比环状结构之间复杂, 较难进行统一描述。但很多非环子结构在大规模分子库中能呈现出一些统计特征, 因此, 本文利用频繁子图挖掘技术对其进行检测和筛选。具体来讲, 为检测一个给定的图数据集
1.3.2 基于交叠子图覆盖最优化的非环结构塌缩 在以塌缩方式处理非环子图结构之前, 还有一个重要的障碍需要克服, 即非环子图之间的重叠问题。由于可用于塌缩的候选非环子图有多种选择, 它们之间存在着相交(两个子图某些部分重叠)或包含(一个子图完全位于另一个子图内部)等交叠现象, 甚至同一类型的子图在同一分子结构图中也会出现多次且会互相重叠(
| 表1 占优子图挑选的算法 Table 1 Dominant subgraph selection |
占优度
为统一表示塌缩后的结构式, 需要引入更具抽象度高层图结构描述方法— — 超图。
定义3:基于化学分子结构式原图
对于有环分子, 以其环图
对于无环分子, 依据表1算法所得占优子图结果, 将
结构式的相似度度量是实现检索目标的最后一步。本文采用了分级匹配的策略:对于一个查询分子, 先计算其与数据集中其他分子在超图级别的最小图编辑距离, 进行初筛, 得到一个候选分子集合, 再在原图级别计算查询分子与候选集中其他分子的最小图编辑距离及其子图同构的数量, 综合评价两者的相似度, 得到更为精确的检索结果。
基于本系统的实验性检索网站为www.pharmki.com。为了评估本系统的检索准确率, 我们将其和维基化学结构检索系统(Wikipedia Chemical Structure Explorer, WCSE)[18]进行了对比, 并通过一些具体的检索实例进一步和WCSE进行直观比较。
本研究实验环境如下:一台配置为3.2 GHz Intel Core i5处理器和16G内存的iMac, 代码由MATLAB R2016b编译。采用的数据集为Wikipedia分子数据集(下文简称为WIKI, 网址为:http://www.cheminfo.org/wikipedia/smiles.txt), 该数据集共有14 676个分子, 每个分子的平均原子数量为41.41, 最大为1 262, 最小为1, 每个分子的平均键数量为42.48, 最大为1 338, 最小为0。
定义2中的参数
WCSE是目前在已公开完整数据集的化学分子结构式检索系统中检索效果最好的, 其所用WIKI数据集可用于对比实验。本文系统与WCSE的对比结果如表2所示, 其中, mean average precision(MAP)、discounted cumulative gain (DCG)、rank-biased precision(RBP)和expected reciprocal rank(ERR)是衡量检索准确性的指标, RBP指标的参数p设定为0.85, 设定检索结果范围为前2~10个, 即表2中top-2到top-10。结果显示, 本文系统在各项指标度量均比WCSE表现更好, 例如, 针对top-10, 上述指标的领先幅度分别为10%、1.41、6.42%、1.32%。具体而言, MAP指标表明本文系统比WCSE能检索到更多的相似结构式; 更高的DCG数值则说明其对结果中的相似结构式排序效果更好。此外, 本文系统在RBP85和ERR指标的更好表现, 则进一步说明其返回的相似结构式结果相比WCSE更符合用户的查询意图。观察表2中各项指标随top-k变化的趋势, 可以发现两者在MAP指标上的表现均逐渐走低, 表明两个系统均倾向于将更相似的结构式优先返回。两者ERR数值趋于稳定, 说明两个系统均能返回满意度较高的结果。但对于DCG和RBP85两个指标, 两系统的差距均逐渐加大, 这表明本文系统在更大范围的检索上表现更优。综上所述, 本文方法不仅能检索到更多相似的分子结构式, 而且检索结果有更好的排序效果, 即优先返回更相似的分子结构式。
| 表2 top-k(2~10)检索结果的MAP(%)、DCG、RBP85(%)、ERR(%)值 Table 2 MAP(%), DCG, RBP85(%), and ERR(%) at |
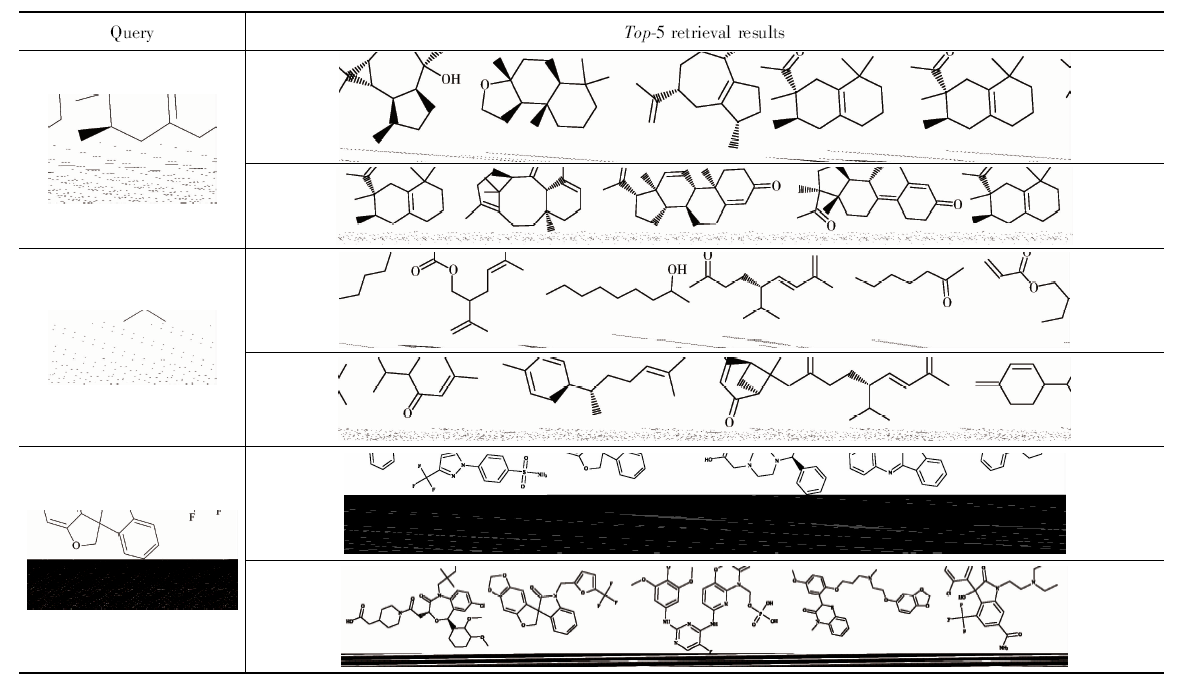
为更直观地比较本文系统和WCSE, 表3展示了一些查询实例及其在两个系统上的检索结果, 设定的检索结果范围为前5个。可见两个系统均能将精确匹配的结果作为第一个候选项返回, 但是在剩余的候选结果中则存在着明显差异。第一个查询分子龙涎酮主要由两个环构成, 本文系统返回了3个同样具有两个环的相似分子, 而WCSE只返回了1个具有两个环的分子, 其余结果和龙涎酮并不相似。第二个查询分子茄酮为无环分子, 主要结构为5-丙基-8-甲基-二烯酮, 本文系统的检索结果均为包含碳链的类似茄酮的无环分子, 而WCSE的返回结果却都是有环分子。第三个查询分子Funapide包含6个环和1个三氟甲基, 本检索系统返回的4个结果均包含1个三氟甲基, 且具有3个或者4个环, 而WCSE的检索结果中只有1个分子具有三氟甲基。
| 表3 WIKI数据集中top-5检索结果对比:本文与WCSE Table 3 Comparison of the top-5 retrieval results from the proposed method and WCSE on WIKI |
针对备受关注的医药信息检索, 结合化学分子结构式的丰富空间结构信息和表征化合物的唯一性, 本文提出了一种基于图结构相似度的分子结构式检索方法。该方法通过智能终端的拍照或手绘实现输入结构式, 以子图匹配和频繁子图挖掘等方法为基础, 结合结构式的结构特性对大图进行多级塌缩, 得到相应的高级抽象表示— — 超图, 其在筛选子图的关键环节采用了基于图同构的子图覆盖分析方法, 并采用不同粒度信息(图编辑距离和子图同构等)实现精确的分子匹配。实验结果显示, 本文方法不仅在多项检索准确率指标均具有优势, 而且直观效果更佳, 表明此方法针对药物分子检索更具有效性, 能为用户提供更为满意的分子检索结果。当然, 在该研究方向上还有许多问题值得进一步探索, 例如, 如何构建大分子的多层级超图、如何设计能够传递三维信息的立体图, 以及如何实现更高效的图匹配等。
The authors have declared that no competing interests exist.
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|