


{kind=link}
{kind=link}
{kind=link}
{kind=link}
嗜铬细胞瘤/副神经节瘤患者 RET、 VHL、 SDHD、 SDHB遗传基因变异的检测
[吴恺1 , 张杨1 , 张红1 , 檀增桓2 , 郭晓蕙1 , 杨建梅1, △  ]
]
]
|
|


目的: 检测嗜铬细胞瘤/副神经节瘤患者的4种与该病相关基因( RET、 VHL、 SDHD、 SDHB)的胚系遗传变异,以了解我国该病患者上述基因变异情况。方法: 以2012年9月至2014年3月就诊于北京大学第一医院,并经病理确诊的嗜铬细胞瘤/副神经节瘤患者为研究对象,共入选12例,其中嗜铬细胞瘤患者11例,副神经节瘤患者1例。提取患者外周血白细胞DNA,采用PCR技术扩增 RET基因的第10、11、13~16外显子, VHL、 SDHD、 SDHB基因的全部外显子,以及各外显子的邻近内含子(±20 bp),并对扩增产物进行测序。比对测序结果及野生型序列,确定基因变异。对1例诊断为多发性内分泌腺瘤病2A型的患者进行家系分析,在家族成员中检测相应基因变异。结果: 共3例患者发现胚系基因变异:1例恶性嗜铬细胞瘤患者存在 SDHB基因c.136C>T(p.R46X)变异;1例多发性内分泌腺瘤病2A型患者存在 RET基因c.1901G>A(p.C634Y)变异及c.2071G>A(p.G691S)/ c.2712C>G(p.S904S)变异,其家系中发现其他5名成员携带上述变异;1例散发、无恶性表现的嗜铬细胞瘤患者存在 RET基因c.2071G>A(p.G691S)/ c.2712C>G(p.S904S)变异。1例患者存在先天性单心室畸形合并嗜铬细胞瘤,其上述4个基因均未发现有临床意义的变异。结论: 本研究在25%(3/12)的嗜铬细胞瘤/副神经节瘤患者中发现了错义或无义胚系基因变异,包括 SDHB基因的c.136C>T(R46X)变异、 RET基因的c.1901G>A(C634Y)变异以及 RET基因的c.2071G>A(p.G691S)/ c.2712C>G(p.S904S)变异等,其中前两种变异具有明确的致病性。本研究验证了上述变异在嗜铬细胞瘤/副神经节瘤患者中的存在,临床工作中应该对嗜铬细胞瘤/副神经节瘤患者开展广泛的遗传基因筛查。
Objective: To analyze the germline variations of genes RET, VHL, SDHD and SDHB in patients with pheochromocytoma and/or paraganglioma and to evaluate variations of these genes in Chinese patients.Methods: Patients who were treated in Peking University First Hospital from September 2012 to March 2014 and diagnosed with pheochromocytoma and/or paraganglioma by pathologists were included in this study. Twelve patients were included in total, of whom 11 had pheochromocytoma, and 1 had paraganglioma. Deoxyribonucleic acid (DNA) was extracted from the leukocytes of peripheral blood of the patients. The exons 10, 11, 13-16 of the RET gene, and all exons of VHL, SDHB and SDHD genes and their nearby introns (±20 bp) were amplified with polymerase chain reactions, and the products were sent to a biotechnology company for sequencing. The sequencing results were compared with wildtype sequences of these genes to identify variations. One of the patients was diagnosed with multiple endocrine neoplasia type 2A. A family analysis was performed in his kindred, and his family members received genetic tests for the related variations.Results: Three patients were found to have germline gene variations. A c.136C>T (p.R46X) variation of the SDHB gene was found in a patient with malignant pheochromocytoma. A c.1901G>A (C634Y) variation, as well as c.2071G>A (p.G691S) and c.2712C>G (p.S904S) variations of the RET gene were found in a patient with multiple endocrine neoplasia type 2A. After a family analysis, five family members of this patient were found to have the same variations. c.2071G>A (p.G691S) and c.2712C>G (p.S904S) variations of the RET gene were also found in a clinical sporadic patient without evidence of malignancy. A patient with congenital single ven-tricle malformation and pheochromocytoma was included in this study, and no variation with clinical significance was found in the four genes of this patient.Conclusion: 25% (3/12) patients with pheochromocytoma or paraganglioma were found to have missense or nonsense germline gene variations in this study, including the c.136C>T (p.R46X) variation of the SDHB gene, the c.1901G>A (C634Y) variation of the RET gene, and c.2071G>A (p.G691S) and c.2712C>G (p.S904S) variations of the RET gene. The former two variations have already been confirmed to be pathogenic. The existence of these variations in Chinese patients with pheochromocytoma and/or paraganglioma was validated in this study, which supports the conclusion that genetic testing is necessary to be generally performed in patients with pheochromocytoma and/or paraganglioma.
嗜铬细胞瘤和副神经节瘤是一类神经内分泌源性肿瘤, 常合称嗜铬细胞瘤/副神经节瘤(pheochromocytoma/paraganglioma, PPGL)。近年研究显示约1/3的PPGL患者具有遗传背景[1], 目前已确定RET、VHL、SDHB、SDHD、SDHC、SDHAF2、SDHA、NF1、TMEM127、MAX等10余个基因的致病性变异可导致PPGL, 其中以RET、VHL、SDHD、SDHB最为常见[1, 2, 3, 4]。
RET基因(Entrez Gene ID:5979)缺陷可导致多发性内分泌腺瘤病2型(multiple endocrine neoplasia type 2, MEN2), 该病分为MEN2A、MEN2B等亚型, 临床可见PPGL、甲状腺髓样癌(medullary thyroid carcinoma, MTC)、甲状旁腺功能亢进等表现。本病有显著的基因型-表型关系:2A型患者缺陷多位于第634密码子, 亦可见于第609、611、618、620、630等密码子; 2B型患者缺陷绝大多数位于第918密码子, 少数位于第883密码子[5]。VHL基因(Entrez Gene ID:7428)缺陷可导致von Hipple-Lindau综合征(VHL综合征), 该病临床分为两个亚型, 其中2型有嗜铬细胞瘤表现, 且有多发倾向, 致病性变异多为错义突变[6]。SDHD基因(Entrez Gene ID:6392)缺陷可致家族性PPGL综合征1型, 临床表现为有多发倾向的PPGL[7, 8], 该病为父源性印迹遗传, 仅来自父亲的缺陷基因可致病[9]。SDHB基因(Entrez Gene ID:6390)缺陷可致家族性PPGL综合征4型, 该病出现恶性肿瘤比例高, 患者预后差[10]。
对PPGL患者开展基因筛查可及时发现潜在的基因背景, 指导对患者及家属的诊治和随访, 有重要的临床意义。因此, 美国内分泌学会《PPGL临床实践指南》建议对所有PPGL患者进行遗传基因筛查[11], 但目前国内临床工作中对PPGL患者的遗传基因筛查尚未广泛开展, 相关研究较少。本研究是一个以初步了解中国PPGL患者遗传基因变异情况为目的的单中心调查性研究。
以2012年9月至2014年3月就诊北京大学第一医院, 经病理诊断为PPGL的患者为研究对象。经北京大学第一医院生物医学研究伦理委员会审查批准(2012[492])并取得患者知情同意后, 留取患者外周静脉血, 提取外周血白细胞DNA。
共入选12例PPGL患者(表1), 包括男性8例、女性4例, 起病年龄12~82岁。其中10例为单发嗜铬细胞瘤; 1例为双侧嗜铬细胞瘤伴MTC(患者P07), 临床诊断为MEN2A, 其父有MTC病史; 1例为多次复发的盆腔副神经节瘤。1例嗜铬细胞瘤(患者P12)和1例副神经节瘤(患者P09)为恶性肿瘤, 其余患者暂未发现转移灶, 无恶性证据。除前述患者P07外均无PPGL或肿瘤综合征家族史。
采用聚合酶链式反应(polymerase chain reaction, PCR)扩增RET基因的第10、11、13、14、15、16外显子[5], VHL基因的全部3个外显子, SDHD基因的全部4个外显子, SDHB基因的全部8个外显子, 及上述外显子的邻近内含子(± 20 bp)。PCR扩增产物由北京天一辉远生物科技有限公司进行测序, 并与野生型序列进行比对分析。对临床诊断MEN2A且有家族史的P07患者, 我们取得其共29名家族成员知情同意后采集了该家族的病史, 并筛查了RET基因变异情况。
入选患者中有3例(P06、P07、P12)检出致病性变异, 占全部的25%, 其中2例为RET基因变异, 1例为SDHB基因变异(表2)。
P06患者检出RET基因c.2071G> A(p.G691S)变异和c.2712C> G(p.S904S)变异(图1)。该患者为女性, 无PPGL或其他肿瘤家族史, 发作性血压升高症状出现于43岁, 45岁时发现右肾上腺直径约4.5 cm占位, 术后病理诊断嗜铬细胞瘤。超声检查于甲状腺及甲状旁腺区未见占位。
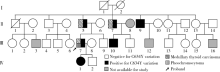
P07患者检出RET基因c.1901G> A(p.C634Y)变异(图2), 同时检出了RET基因c.2071G> A(p.G691S)和c.2712C> G(p.S904S)变异。患者为男性, 发作性血压升高出现于25岁, 27岁时发现双侧肾上腺占位, 左侧7.46 cm× 4.95 cm, 右侧6.08 cm× 3.21 cm; 甲状腺超声提示双侧甲状腺占位; 降钙素> 900 ng/L。术后病理诊断嗜铬细胞瘤及MTC, 结合基因检测诊断为MEN2A。患者父亲亦患MTC, 提示患者父系可能为一个MEN2A家系。我们对该家系共29名亲属进行了基因筛查和临床病史采集(图3), 筛查中先证者1名叔父确诊为MTC, 共发现5名亲属(Ⅱ 5、Ⅱ 7、Ⅱ 9、Ⅲ 10、Ⅳ 1)存在C634Y变异和G691S/S904S变异, 证实该家系为MEN2A家系。此外, 与该家族无血缘关系、无临床表现的Ⅱ 14亦携带G691S/S904S变异。
| 表1 患者一般情况 Table 1 General condition of patients |
| 表2 基因检测结果 Table 2 Result of the gene testing |
P12患者检出SDHB基因c.136C> T变异(图4), 该变异使第46位密码子CGA改变为终止密码子UGA(p.R46X), 为无义突变。该患者为男性, 无PPGL家族史, 42岁出现高血压, 药物控制不良, 44岁起出现发作性血压升高症状。45岁时运动后出现心功能不全, 影像学检查发现右肾上腺约12.3 cm× 12.2 cm巨大占位; 超声心动图提示心肌病变、左心增大, 左心室射血分数(Teich法)33%, 临床诊断儿茶酚胺心肌病可能; 131I-间碘苄胍(metaiodobenzylguanidine, MIBG)显像示右肾上腺部位不规则巨大异常放射性浓聚区。手术切除右肾上腺区15 cm× 11 cm× 10 cm肿物及肿物粘连部分右肾, 病理诊断为恶性嗜铬细胞瘤。
P05为先天性单心室合并嗜铬细胞瘤患者。患者出生后确诊为先天性单心室畸形, 12岁时发现嗜铬细胞瘤。本研究中未发现该患者SDHD、SDHB、VHL、RET遗传基因致病性变异。
 | 图1 患者P06 RET基因变异情况Figure 1 Variation of the RET gene of patient P06A, c.2071G> A variation in the RET gene of patient P06; B, c.2071 in wildtype RET gene; C, c.2712C> G variation in the RET gene of patient P06; D, c.2712 in wildtype RET gene. |
 | 图2 患者P07 RET基因变异情况Figure 2 Variation of the RET gene of patient P07A, c.1901G> A variation in the RET gene of patient P07; B, c.1901 in wildtype RET gene; C, c.2071G> A variation in the RET gene of patient P07; D, c.2712C> G variation in the RET gene of patient P07. |
 | 图3 患者P07家系图Figure 3 Pedigree of patient P07 |
 | 图4 P12患者SDHB基因变异情况Figure 4 Variation of the SDHB gene of patient P12A, c.136C> T mutation in the SDHB gene of patient P12; B, c.136 in wildtype SDHB gene. |
本研究入选的12例PPGL患者中3例检出了遗传基因致病或可能致病的变异, 占总数的25%, 如除去临床有家族性MEN2A表现的P07, 则在散发表现的患者中有18%发现遗传基因变异。Neumann等[6]在德国和波兰的271例散发性嗜铬细胞瘤患者中进行的筛查提示, 约24%患者有遗传背景; Waldmann等[3]对德国36例嗜铬细胞瘤患者进行研究, 其中有13例(36%)发现致病性变异, 包括10例临床确诊为MEN2的患者, 如仅统计临床散发表现的患者, 则存在致病性变异的比例为11.5%。本研究限于规模和时间, 入选病例较少, 无法与上述类似研究进行统计学比较, 但研究结果仍显示我国PPGL患者中存在遗传背景的比例值得重视, 开展遗传基因筛查具有重要临床意义。
本研究在P12患者的SDHB基因发现了c.136C> T(p.R46X)变异, 是一个已证实的致病性变异, 此前国内曾有2例报道[12, 13]。该患者临床表现为右肾上腺恶性嗜铬细胞瘤, 131I-MIBG显像未发现多灶性表现, 临床未发现副神经节瘤。Neumann等[7] 对32例SDHB缺陷基因携带者的研究显示, 该类患者约34%存在恶性PPGL, 显著多于SDHD缺陷基因携带者及非SDHB/SDHD基因缺陷相关PPGL患者。Benn等[8]对SDHB基因缺陷家系的研究显示, 49例患者中22%为恶性肿瘤患者。Dhir等[14]在60例PPGL中发现11例SDHB基因缺陷, 均为恶性肿瘤患者。上述研究均提示, SDHB基因缺陷患者出现恶性肿瘤比例较高, P12患者的临床表现与此结论相符。Benn等[8]认为SDHB基因缺陷导致PPGL的外显率较低, 但Jochmanova等[15]近期的研究提示, R46X变异较其他常见SDHB致病性变异具有较早的起病时间(外显率达50%的年龄为38岁), 因此, 在PPGL患者中筛查SDHB变异情况对判断预后等具有较大意义。
P07患者的RET基因C634Y变异是一个已证实的MEN2A致病性变异, 证实了患者的临床诊断。对该家系成员的RET基因检查、降钙素测定及临床随访发现了先证者及其父之外的1例MTC患者, 确定了先证者之外的5例RET基因C634Y变异携带者。国内外研究显示绝大多数MEN2A患者致病性基因变异位于第634位密码子上[16, 17, 18, 19, 20], 本研究亦印证了这一结论。除先证者合并双侧嗜铬细胞瘤外, 该家系中主要临床表现均为MTC, 而早期发现和治疗MTC对于改善预后至关重要。美国甲状腺协会在指南中建议必要时对MEN2高危致病性变异携带者施行预防性甲状腺切除术[5], 相关国内研究亦支持必要的预防性手术[21], 故开展PPGL患者的RET基因检测, 对于潜在的MEN2患者诊治具有重要意义。
P07患者亦存在RET基因的c.2071G> A(p.G691S)/c.2712C> G(p.S904S)变异(以下简称G691S/S904S), 其母(图3中Ⅱ 6)未发现该变异, 而家系中其他C634Y携带者均存在G691S/S904S, 提示P07患者的C634Y和G691S/S904S均来自于父亲, 两者有连锁关系。P06患者也发现了RET基因的G691S/S904S变异, 未发现其他致病性变异, 其临床表现为单侧嗜铬细胞瘤, 无甲状腺肿瘤, 无家族史。G691S/S904S变异的高度连锁性已由Gil等[16]报道, 且被多个其他研究证实, 但该变异与MEN2的关系尚未明确。Siqueira等[17]的研究发现, G691S/S904S在MEN2患者中出现的频率与健康人群中无明显差异, 提示该变异可能无法单独导致MEN2, 但G691S/S904S可能在MEN2起病过程中有调节作用。同时存在G691S/S904S和某些其他RET基因单核苷酸多样性(如c.2307T> G, p.L769L或c.2508C> T, p.S836S)或G691S/S904S纯合子的MEN2患者, 都表现出MTC和嗜铬细胞瘤起病更早的倾向[17, 22]。P07患者确诊年龄较早(27岁), 但此趋势尚需进一步证实。对该家系的随访可能有助于进一步了解G691S/S904S对MEN2A临床表现的影响。在G691S/S904S对MTC的致病作用方面, Figlioli等[23]的研究认为该变异对MTC无明显致病效应, 而Lantieri等[24]则认为其致病效应不高于一个隐性致病基因, 推测该效应可能是通过G691S产生的, 但目前尚无研究显示G691S/S904S对PPGL的致病作用。结合上述研究结果, 我们推测G691S/S904S可能不会导致PPGL, 或其致病效应较弱, 因而在小规模研究中无法明确, 更大规模的研究数据可能有助于明确这一推测。
本研究发现1例先天性单心室合并嗜铬细胞瘤患者(P05), 查阅文献, 国外曾有5例单心室合并嗜铬细胞瘤的报道[25, 26, 27, 28], 但国内尚未见类似病例报道。单心室畸形为罕见病, 而该病与另一罕见病PPGL的合并可能提示了两者之间的联系。阿部等[27]结合PPGL的低氧机制假说, 提出单心室畸形导致长期的右向左分流, 使体循环动脉血氧饱和度下降, 组织长期处于低氧状态, 导致低氧反应通路持续激活, 可能促使了肿瘤的形成。根据此假说, 患者可能不存在SDHD、SDHB胚系基因致病性变异, 而存在因长期低氧产生的肿瘤体细胞基因改变。本研究在P05患者遗传基因中未发现SDHD、SDHB基因的胚系致病性变异, 符合上述推论, 但进一步验证该假说需要对肿瘤体细胞相关基因变异、表达的情况进行研究。
综上所述, 本研究在25%(3/12)PPGL的患者中发现了致病性或可能致病的遗传基因变异。1例恶性嗜铬细胞瘤患者存在SDHB基因c.136C> T, p.R46X无义突变; 1个MEN2A家系存在RET基因c.1901G> A, C634Y变异以及该基因的G691S/S904S变异; 1例单发嗜铬细胞瘤、暂无恶性表现的患者存在RET基因G691S/S904S变异。尽管本研究入选病例数较少, 但研究结果仍表明上述变异可能存在致PPGL作用, 显示出基因筛查对PPGL患者的重要性以及家族疾病监测的意义, 应在PPGL患者中广泛开展基因筛查工作。
(本文编辑:赵 波)
The authors have declared that no competing interests exist.
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|