


{kind=link}
{kind=link}
{kind=link}
皮肤浆细胞增多症1例
[张思, 李文海, 赵琰, 蔡林△  ]
]
]
|
|


SUMMARY A collection of plasma cells in the skin can represent a broad spectrum of disease entities. Secondary syphilis, primary cutaneous plasmacytoma, primary cutaneous plasmacytosis, cutaneous lymphoid hyperplasia and nodular amyloidosis are considered possible differential diagnoses. The primary cutaneous plasma cell disorders can range from malignant to benign plasma cell neoplasms. The malignant conditions are neoplastic diseases having monoclonal proliferations, rapid progression and fatal outcome while the benign plasma cell disorders usually show polyclonality, chronicity and benign process, including plasmacytosis. We present a case of cutaneous plasmacytosis. The patient was a 34-year-old man, presented with disseminated reddish-brown plaques and nodules on the right side of the hips, inguinal groove, and the thigh. Histopathologically, mature plasma cells perivascular infiltrates were observed mainly in the dermis. Polyclonality of infiltrating plasma cells with coexistence of both kappa and gamma chain-positive cells demonstrated with immunohistochemistry, as well as CD20+++, CD38++++, CD79a++++, CD138++, Ki67<30%. The diagnosis, cutaneous plasmacytosis, was established by the pertinent laboratory findings. Primary cutaneous plasmacytosis was an uncommon reactive lymphoplasmacytic disorder of uncertain etiology. Cutaneous plasmacytosis is a rare disease characterized by peculiar multiple eruptions and hyper gamma globulinemia. It has been mainly described in patients of Japanese descent, with only few reports in Caucasians and Chinese, although information concerning the disorder was limited to individual case reports. Cutaneous plasmacytosis is a rare disorder, which is characterized by multiple red to dark-brown nodules and plaques on the trunk and usually associated with polyclonal hyper gamma globulinaemia. Primary cutaneous plasmacytosis or cutaneous plasmacytosis was thought to be a reactive process with unknown etiology. Histologically, lesions contain dense perivascular infiltration of mature polyclonal plasma cells without any atypia, in the dermis and subcutaneous fat. The clinical course is chronic and benign without spontaneous remission. Available treatments for cutaneous plasmacytosis include psoralen ultraviolet A radiotherapy, systemic chemotherapy and intralesional steroid injection. The patient with cutaneous plasmacytosis in this report was treated with tacrolimus ointment and psoralen ultraviolet A.
皮肤合并系统性浆细胞增多症(cutaneous and systemic plasmacytosis, CSP)是一种以皮肤或多系统成熟浆细胞浸润为特征的良性疾病, 最常累及皮肤、淋巴结、骨髓, 发病率较低, 亚洲人多见, 男性发病率稍高, 典型的发病年龄为20~55岁。该病的特征为浆细胞多克隆增殖, 皮肤浆细胞增多症(cutaneous plasmacytosis, CP)仅累及皮肤, 较为罕见, 皮损常表现为棕红色斑块, 局部外用糖皮质激素或他克莫司、长波紫外线照射有一定疗效。北京大学人民医院皮肤科收治1例皮肤浆细胞增多症, 现报道如下。
患者, 男性, 34岁, 因“ 右侧臀部、腹股沟、股内侧红斑10个月” 于2016年5月23日于我院就诊。患者10个月前无明显诱因右侧臀部出现钱币大小红色斑片, 边界不清, 伴瘙痒, 未诊治。皮疹面积逐渐扩大, 皮疹数量增多, 部分融合至巴掌大小红色斑块, 右侧腹股沟及股内侧亦出现类似皮疹, 经自行外用皮炎平治疗, 未见好转, 多次于外院就诊行皮肤组织病理活检, 诊断为皮肤浆细胞增多症。患者无发热、咳嗽、咳痰、恶心、呕吐、腹痛、腹泻、头晕、头痛等症状, 大小便正常, 体重无明显改变。既往体健, 家族无类似病史。
体格检查:系统检查未见异常, 全身浅表淋巴结未触及肿大。皮肤科检查可见右侧臀部、腹股沟、股内侧红色浸润性斑块、结节, 大小不一, 边界不清, 部分融合(图1), 皮疹表面散在破溃、结痂, 无鳞屑。
皮肤组织病理学检查:表皮正常, 真皮全层及皮下组织血管、附属器周围有大量成熟浆细胞和较多淋巴细胞浸润, 可见少量嗜酸性粒细胞, 未见异型细胞(图2); 免疫组织化学染色:CD20+++、CD38++++、CD79a++++、CD138++、Ki67< 30%、血免疫球蛋白κ 轻链++、λ 轻链+(图3)。
实验室及辅助检查:血常规及尿常规正常, 尿本周氏(Bence-Jones)蛋白阴性。血生化检查未见肝、肾功能异常, 血IgA、IgD、IgM、IgG及补体C3、C4正常, γ 球蛋白(15.2%)正常, 血免疫球蛋白κ 、λ 轻链正常。类风湿因子及抗链球菌溶血素O阴性, C反应蛋白未见异常, 梅毒螺旋体血凝试验阴性, 快速血浆反应素环状卡片试验阴性。骨髓细胞学检查、骨髓穿刺活检、甲状腺功能及凝血功能均未见异常。胸部X线片提示双肺纹理稍增强, 心电图未见异常, 腹部彩超提示肝、胆、胰、脾及双肾均未见明显异常。
诊断:皮肤浆细胞增多症。
皮肤浆细胞增多症是一种少见的多克隆浆细胞增生性疾病, 多见于亚洲人, 特别是日本人[1], 好发年龄为20~62岁, 平均发病年龄37岁, 男女发病比例约为1 :0.6[2]。皮肤浆细胞增多症的发病机制至今仍不明确, 有学者认为其为一种反应性浆细胞病[2]。大多数患者血清白细胞介素6(interleukin-6, IL-6)水平升高, IL-6可以诱导B细胞增殖并分化成浆细胞, 提示IL-6的升高在本病的发病机制中起着重要的作用[3]。有研究推测该病可能类似于肥大细胞增生症的发病机制, 即由于调控浆细胞分化的信号传导分子发生突变, 原癌基因C-kit点突变, 引起KIT(一种酪氨酸激酶)活化, 从而阻止了浆细胞的凋亡[4]。因多见于日本人, 因此怀疑特定环境中的感染因素也在本病的发病中起着一定作用[4]。
浆细胞增多症分为皮肤浆细胞增多症和皮肤合并系统性浆细胞增多症两种类型, 前者仅累及皮肤, 临床罕见; 后者除皮肤以外, 常累及两个或两个以上的器官, 如淋巴结、骨髓、肺等, 除典型皮疹以外, 常伴有高γ 球蛋白血症、淋巴结肿大及骨髓浆细胞浸润等表现, 多数患者不伴有全身症状, 少数可有发热、呼吸道症状等。
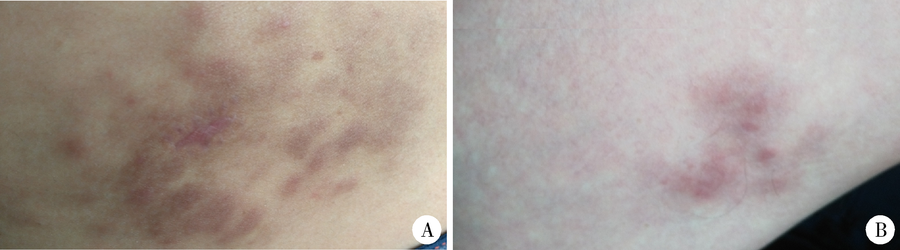 | 图1 皮肤浆细胞增多症患者右侧臀部及股内侧皮疹Figure 1 Lesions on the right side of the hips, and the thigh of the patient with cutaneous plasmacytosisA, disseminated reddish-brown plaques and nodules with a unclear boundary on the right side of the hips; B, multiple fution brown plaques on the right thigh. |
 | 图2 右侧大腿皮疹皮肤组织病理表现(HE × 100)Figure 2 Skin tissue pathology of the lesions on the right thigh (HE × 100)Dense perivascular infiltration in the upper and mid dermis. Higher magnification reveals numerous plasma cells mixed with lymphocytes andhistiocytes. |
 | 图3 右侧大腿皮疹免疫组织化学染色Figure 3 Immunohistochemistry of the lesions on the right thighA, numerous plasma cells and lymphocytes with expression of CD20 (+++); B, plasma cells with expression of CD38 (++++); C, plasma cells with expression of CD138 (++); D, plasma cells and lymphocytes with expression of CD79a (+++). |
皮肤浆细胞增多症的皮肤组织病理表现为真皮血管及附属器周围有大量成熟浆细胞浸润, 细胞无异型, 可伴有淋巴细胞及组织细胞浸润。免疫组织化学标记浆细胞同时表达κ 和λ 轻链(κ /λ 比值介于1/10~10为多克隆增生)[5], 成熟浆细胞还表达CD38、CD138和CD79a, 而CD20一般为阴性。淋巴结活检示皮质及髓质区有成熟浆细胞增生, 骨髓穿刺可见成熟浆细胞增多, 无异型浆细胞。
在组织学上本病需要与其他浆细胞增生性疾病鉴别:(1)单克隆浆细胞增生性疾病:主要有原发性皮肤浆细胞瘤, 组织病理可见异型浆细胞, 并可以观察到成熟浆细胞和不成熟浆细胞之间的过度状态, 有时还可见Russell小体和Dutcher小体, 此为恶性肿瘤, 常为多发性骨髓瘤的皮肤表现。(2)多克隆浆细胞增生性疾病:主要为多中心Castleman病, 与人疱疹病毒8型感染明显相关, 表现为无痛性淋巴结肿大, 伴长期发热、乏力、消瘦、贫血、肝脾肿大, 很少累及皮肤, 该病临床表现严重, 病程进展快, 预后差。(3)感染:主要见于梅毒、真菌感染等, 可行梅毒血清学试验予以鉴别。
本例患者临床表现为臀部、腹股沟及股内侧棕红色斑块、斑片; 组织病理见真皮血管及附属器周围成熟浆细胞浸润, 未见异型细胞, 免疫组织化学染色CD38++++、CD79a++++、CD138++、血免疫球蛋白κ 轻链++、λ 轻链+。其临床皮损及组织病理均符合皮肤浆细胞增多症的典型表现, 且血清γ 球蛋白不高, 除皮疹外暂无其他系统受累的表现, 故可排除系统性浆细胞增多症, 诊断为皮肤浆细胞增多症。
皮肤浆细胞增多症至今没有标准的治疗方案, 且疗效不一。目前的治疗方法有系统及局部或外用糖皮质激素, 应用抗CD20单抗系统化疗等[4, 6]。局部放疗、补骨脂长波紫外线照射、光动力也有一定疗效。有报道局部外用0.1%他克莫司软膏或1%吡美莫司乳膏可成功治愈本病[7], 也有报道口服曲安西龙联合阿维A 1个月, 同时肌肉注射重组人干扰素9个月, 皮疹即可完全消退, 且无复发[8], 但仅为个案。
皮肤浆细胞增多症进展缓慢, 很少自行消退, 但预后大部分良好。本例患者仍在随访中, 目前其皮疹面积较前缩小, 颜色变暗。
(本文编辑:赵 波)
The authors have declared that no competing interests exist.
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|