
{kind=link}
{kind=link}
{kind=link}
{kind=link}
氯喹抑制肾癌细胞活性促进舒尼替尼诱导的细胞凋亡
[孙静1, 2 , 宋卫东3 , 闫思源2 , 席志军3, △  ]
]
]
|
|


目的: 研究氯喹(经常用于抑制自噬体与溶酶体的融合,是一种晚期自噬的抑制剂)是否能抑制肾癌细胞的增殖以及对舒尼替尼(sunitinib, ST)诱导的细胞凋亡的影响。方法: 以肾癌细胞系786-O和ACHN为模型,利用3-(4,5-二甲基)-5-(3-羧甲基苯环)-2-(4-硫基苯)-2H-四唑盐复合物检测法[3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt, MTS]观察氯喹对细胞活性的影响;透射电子显微镜、免疫杂交等手段检测氯喹影响ST诱导的凋亡、自噬的情况。应用早期自噬抑制剂3-甲基腺嘌呤(3-methyladenine, 3-MA)、RNA干扰技术敲降自噬相关蛋白Ulk1 (unc-51-like kinase 1)和微管相关蛋白1轻链3融合蛋白(microtubule associated protein 1 light chain 3 fusion protein, LC3)确定早期自噬对ST引起的细胞凋亡的影响。结果: 氯喹和ST均可以抑制786-O和ACHN细胞的增殖,氯喹可促进ST诱导的细胞凋亡。与氯喹不同,抑制早期自噬减少了ST诱导的细胞凋亡和活性丢失。结论: 舒尼替尼既可以引起细胞自噬,也可以促进细胞的凋亡;早期、晚期自噬对ST诱导的细胞凋亡作用不同,氯喹可以作为一种潜在的抗肾癌药物进行相关的临床研究。
Objective: To determine whether chloroquine (CQ), an often used inhibitor of late autophagy and autophagosome/lyosome fusion, can inhibit proliferation of renal carcinoma cells and investigate its effect on sunitinib (ST)-induced apoptosis.Methods: Renal carcinoma cell line 786-O and ACHN had been used as cellular model and 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt (MTS) assay was carried out to detect the cell viability in response to CQ or ST treatment. Both transmission electron microscope and immunoblotting had been employed to observe apoptotic and autophagic process. To examine the involvement of autophagy in ST-dependent apoptosis, autophagy had been inhibited either chemically or genetically via utilizing autophagy inhibitor or specific small interference RNA (siRNA) targeted to either Ulk1(unc-51-like kinase 1) or LC3 (microtubule associated protein 1 light chain 3 fusion protein), two essential autophagic proteins.Results: Both ST and CQ induced cell viability loss, indicating that either of them could inhibit renal cancer cell proliferation. Clone formation experiments confirmed the aforementioned results. Furthermore, the combined ST with CQ synergistically promoted the loss of cell viability.By transmission electron microscopy and immunoblotting, we found that the ST induced both autophagy and caspase-dependent apoptosis. While 3-MA, an early autophagy inhibitor, reduced the ST-induced cleavage of poly (ADP-ribose) polymerase-1 (PARP-1), a substrate of caspase 3/7 and often used marker of caspase-dependent apoptosis, CQ promoted the ST-dependent PARP-1 cleavage, indicating that the early and late autophagy functioned differentially on the ST-activated apoptotic process. Moreover, the knock down of either Ulk1 or LC3 decreased the ST-caused apoptosis.Interestingly, we observed that rapamycin, a specific inhibitor of mTOR (mammalian target of rapamycin) and an inducer of autophagy, also showed to inhibit cell viability and increased the cleavage of PARP-1 in the ST-treated cells, suggesting that autophagy was likely to play a dual role in the regulation of the ST-induced apoptosis.Conclusion: ST activates both apoptotic and autophagic process in renal carcinoma cells. Although autophagy precedes the ST-induced apoptosis, however, early and late autophagy functions differentially on the apoptotic process induced by this compound. Additionally, ST can coordinate with the inducer of autophagy to inhibit the cell proliferation. Further research in this direction will let us illuminate to utilize CQ as a potential drug in the treatment of renal carcinoma.
肾细胞癌(renal cell carcinoma, RCC)起源于肾小管上皮, 是除膀胱癌以外最常见的泌尿系恶性肿瘤, 约90%~95%的原发性肾恶性肿瘤为肾癌[1]。因其具有多耐药基因, 肾癌对化疗放疗均不敏感。近年来, 由于基础研究的进展, 有很多靶向药物陆续上市用于临床治疗肾癌, 如索拉非尼(sorafenib)、舒尼替尼(sunitinib, ST)等[2]。这些药物的应用可以有效延长晚期肾细胞癌患者的肿瘤特异生存时间(cancer special survival)和总生存时间(overall survival)[3]。以往的研究表明, 舒尼替尼可以抑制多种受体酪氨酸激酶(如VEGFR1、VEGFR2、VEGF3)、血小板衍生内皮生长因子受体(PDEGFRα 、PDEGFRβ )、c-kit受体、FMS样酪氨酸激酶3(fms-like tyrosine kinase, FLT3)等[4]。本课题组之前的研究发现[5], ST能通过mTOR信号通路诱导肾癌细胞ACHN的自噬。
自噬是真核生物中普遍存在的现象, 根据其形态将其分巨自噬、微自噬和分子伴侣介导的自噬[6]。根据降解的目标, 自噬又可以分为核自噬、线粒体自噬、过氧化物酶体自噬[6]。过度的自噬可以发展成为自噬性细胞死亡, 命名为第二类程序性细胞死亡[7]。随着研究的不断深入, 与凋亡类似, 自噬也成为肿瘤治疗的靶点之一。自噬有多种信号通路的调节, 其中主要有两种:腺苷酸活化激酶(AMP-activated protein kinase, AMPK)信号通路和磷脂酰肌醇3-激酶/蛋白激酶B/雷帕霉素靶蛋白(phosphatidylinositol 3-kinase /protein kinase B/mammalian target of rapamycin, PI3K/Akt /mTOR)信号通路[8, 9]。雷帕霉素(rapamycin)是mTOR的特异性抑制剂, 由于肾癌细胞往往伴随着mTOR信号的增加, 因此, 雷帕霉素在临床上可以用于肾癌的治疗[10]。丝苏氨酸蛋白激酶Atg1在哺乳动物细胞中的同源蛋白为Ulk1(unc-51-like kinase 1)[11], 其不同磷酸化位点的磷酸化水平影响其对自噬的调节。Ulk1的555位丝氨酸被AMPK磷酸化而诱导细胞自噬的发生[12], 而757位丝氨酸被mTOR磷酸化后抑制细胞自噬的诱导[13]。此外, Ulk1作为吞噬泡组装位点的支架蛋白介导自噬过程的起始[14]。LC3是一种类泛素蛋白, 可以结合磷脂酰乙醇胺形成膜定位的LC3-Ⅱ [15]。LC3-Ⅱ 是完整自噬体唯一稳定的标记物, 常用于自噬的检测。通过检测细胞内的LC3-Ⅱ 虽然能够间接反映自噬体的含量, 但是单纯LC3-Ⅱ 的增加/减少并不能如实反映自噬的增强/减弱, 而应该检测自噬流[6]。常用的自噬抑制剂有3-甲基嘌呤(3-methyladenine, 3-MA)和氯喹(chloroquine, CQ), 前者抑制早期自噬, 而CQ是常用的晚期自噬抑制剂, 也常被用于检测自噬流[6]。
早在1944年, 氯喹就用于临床上治疗疟疾, 之后又发现其可以用于其他疾病的治疗, 随着自噬的研究深入, 人们开始认识到氯喹用于肿瘤治疗的潜在价值[16, 17], 但是就目前情况而言, 有关氯喹作用与肾癌的研究还比较少见, 其是否可以与舒尼替尼协同抑制肾癌增殖尚未见报道, 在之前的研究基础上, 本课题组就氯喹是否抑制肾癌细胞活性以及影响舒尼替尼引起的细胞凋亡展开研究, 以期为临床肾癌治疗提供实验数据。
舒尼替尼(S126061) 购自美国Aladdin公司; 3-甲基腺嘌呤(3-methyladenine, M9281)、氯喹(chloroquine diphosphate salt, C6628)、雷帕霉素(R0395)、LC3B抗体均购自美国Sigma Aldrich公司; PARP-1(9542)、Ulk1 (8054)、p44/42 MAPK (total-Erk1/2, 9102)抗体均购自美国Cell Signaling Technology公司。Ulk1(sc-44182)、LC3(sc-43390)特异性和对照siRNA(sc-37007)均购自美国Santa Cruz Biotechnology公司。Actin(TA-09)抗体购自北京中杉金桥公司。
CO2细胞培养箱购自日本Panasonic SANYO公司, 小型台式离心机购自美国Thermo公司, 台式冷冻高速离心机购自美国Sigma-Aldrich公司, -20 ℃低温冰箱购自中国海尔公司, -80 ℃低温冰箱购自德国Heraeus公司, AL204 电子天平购自美国Mettler Toledo公司, 正置荧光显微镜(Axio Imager A1)购自德国Zeiss公司, Mini Trans-Blot电转仪购自美国Bio-RAD公司, 酶标仪购自瑞士TECAN公司, AE-6500型电泳槽购自日本ATTO 公司, JY600C恒压恒流电泳仪购自中国北京君意东方电泳设备有限公司, MilliQ plus超纯水系统购自美国Millipore公司。
DMEM培养基和胎牛血清购自美国GIBCO公司, 青链霉素混合物和胰蛋白酶消化液购自南京凯基生物科技发展有限公司, PVDF膜购自美国Amersham公司, 发光液购自美国Thermo公司。
786-O和ACHN 购自美国ATCC, 细胞用含10%(体积分数)胎牛血清和1%(质量分数)青链霉素双抗的DMEM培养基, 置于37 ℃、5%(体积分数)CO2细胞培养箱中进行培养, 当细胞生长密度达到80%时弃去旧培养基, 用PBS洗一遍, 用胰酶消化贴壁细胞, 然后以1 000 r/min离心3 min后收集细胞, 弃上清液, 加入新鲜培养基重悬。将细胞分到六孔板内, 37 ° C 5% CO2细胞培养箱中进行培养过夜, 使细胞生长至60%~70%密度, 加药处理细胞。
在准备siRNA干扰实验12 h前, 把所需细胞以上述方法按30%密度接种于培养皿中。依据转染试剂说明书将siRNA和转染试剂分别加入无血清DMEM培养基中, 静置5 min后混合, 再次静置20 min, 之后将混合好的DMEM培养基加入新培养基(含血清, 不含抗生素)的细胞培养皿中, 转染siRNA 48 h后将细胞按照70%左右的密度分到六孔板中, 继续培养12 h后按需要浓度加入舒尼替尼和其他化合物, 处理指定时间。
将786-O细胞接种于直径10 cm的培养皿中, 于37 ℃ 5% CO2细胞培养箱中培养过夜, 至细胞密度达到60%~70%。换新鲜培养基, 按8 μ mol/L终浓度加入舒尼替尼或DMSO处理4 h。完成处理时间后用不含EDTA胰酶消化细胞, 4 ℃ 、1 000 r/min 离心5 min后收集细胞, 冷PBS洗涤两遍。加入3%(体积分数)戊二醛溶液, 4 ℃固定过夜, 送样检验。
药物按指定时间处理细胞后, 弃去培养液, 用冷PBS洗一遍, 加入一定量的TGH细胞裂解液[组成: 1 mL母液[1%(体积分数)Triton X-100, 10%(体积分数)甘油, 50 mmol/L Hepes pH 7.4], 2.5 mol/L NaCl 20 μ L, 0.5 mol/L EGTA/EDTA 10 μ L, 0.1 mol/L NaF 10 μ L, 0.1 mol/L PMSF 20 μ L, 1 mol/L DTT 2 μ L, 0.5 mol/L Na3VO4 2 μ L, 蛋白酶抑制剂1 μ L], 然后用细胞刮铲均匀收集培养皿所有部位的细胞至EP管中。加入TGH使用量1/2体积的3× 上样缓冲液, 96 ℃煮样30 min后, 室温13 000 r/min 离心15 min后收集上清液。等量样品经过8%(或13.5%) SDS-PAGE分离后, 将胶内蛋白质转移至PVDF膜上。将转有蛋白质的PVDF膜按如下次序处理: 5%(质量分数)奶粉封闭液室温封闭60 min, 一抗4 ℃孵育过夜, 洗掉未结合一抗, 二抗室温孵育60 min, 经适当漂洗后涂抹发光液, 并于暗室曝光洗片, 最后将底片扫描并进行分析。
将5 000~10 000个细胞均匀接种于96孔板的每个孔中, 体积为100 μ L, 培养过夜后换无酚红DMEM(含10%胎牛血清)培养基。每组3个孔做为平行对照, 加入指定浓度的舒尼替尼, 37 ℃ 5% CO2细胞培养箱中继续培养到指定的时间。向每孔中加MTS和PMS(20 :1)混合溶液20 μ L, 持续培养并使用酶标仪于492 nm 波长测定各孔光密度值(每30 min测量1次, 直到对照组的光密度值达到0.6), 并记录结果。
将786-O细胞按照900个/盘接种到6 cm培养皿中, 加入2 μ mol/L的ST处理, 37 ℃ 5% CO2细胞培养箱中继续培养1周。吸弃培养基, 用PBS清洗1次, 加入2 mL吉姆萨(Glemsa)染液, 过夜室温孵育。去除染液, 清水清洗1次后, 室温干燥后照相。
使用SPSS 11.0软件, 计量资料以均数± 标准差表示, MTS实验结果符合正态分布, 以单因素方差分析和Student-Newman-Keuls (S-N-K)法两两比较分析, P< 0.05认为差异有统计学意义。
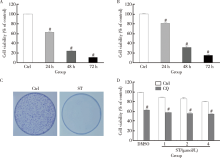
氯喹1944年开始应用于临床治疗, 最初用于治疗疟疾, 以后的用途逐步扩大。 1951年, 氯喹在临床上用于治疗类风湿关节炎, 且具有一定的疗效。随着对细胞自噬的深入认识, 人们普遍认为自噬可以作为肿瘤治疗的一个靶点[18]。由于氯喹能抑制自噬体与溶酶体的融合从而抑制晚期自噬, 因此, 很多学者将氯喹用于肿瘤治疗的临床研究工作。MTS实验表明氯喹可以抑制肾癌细胞786-O的增殖, 其抑制活性具有时间依赖的特点(图1A)。本课题组的前期结果显示舒尼替尼作为肾癌二线靶向治疗药物, 对肾癌细胞ACHN具有杀伤作用。从图1B中可以看出, 舒尼替尼可以抑制ACHN细胞的活性。克隆形成实验进一步显示舒尼替尼具有抑制肿瘤细胞增殖的作用(图1C)。联合应用氯喹促进了舒尼替尼引起的细胞活性的丢失, 二者间具有协同抑制肿瘤的作用(图1D)。
 | 图1 氯喹协同促进舒尼替尼诱导的细胞活性丢失Figure 1 CQ promotes ST-induced cell viability lossA and B, 786-O and ACHN cells were treated with chloroquine (CQ) or sunitinib (ST, 8 μ mol/L) upon to 72 h, and then the cell viability was analyzed by MTS, respectively; C, colony growth assays were performed in 786-O cells with ST (2 μ mol/L) for one week; D, after treatment with 0-4 μ mol/L ST in the presence or absence of CQ (20 μ mol/L), cell viability of 786-O cells were measured by MTS. DMSO, dimethyl sulfoxide. # P < 0.01 vs. control (Ctrl). |
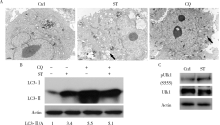
观察细胞自噬体最好的方法就是透射电子显微镜[6], 通过电子显微镜观察发现用舒尼替尼(4 μ mol/L, 4 h)和氯喹(20 μ mol/L, 2 h)处理786-O细胞均可以观察到自噬体的增加(图2A, 箭头指示自噬体结构)。与舒尼替尼处理不同, 氯喹处理的细胞泡状结构内的物质较多, 预示自噬体内容物的降解遇到了障碍。LC3以LC3-I和LC3-Ⅱ 两种形式存在, 前者与自噬体膜上的磷脂酰乙醇胺结合成为LC3-Ⅱ , 在自噬发生时, LC3-I脂化形成LC3-Ⅱ , 从而使后者含量增加[19]。氯喹由于能阻断自噬体与溶酶体的融合, 因此抑制了LC3-Ⅱ 降解, 由此可以用来判断动态自噬发生情况。利用免疫杂交, 发现在氯喹存在的情况下, 舒尼替尼引起的LC3-Ⅱ 会继续增加, 说明舒尼替尼可引起786-O细胞的自噬(图2B)。自噬相关蛋白Ulk1的555位丝氨酸磷酸化有利于自噬的起始, 在舒尼替尼的处理下, 786-O细胞内的555位磷酸化Ulk1明显增加, 表明舒尼替尼可诱导786-O细胞自噬的发生(图2C)。
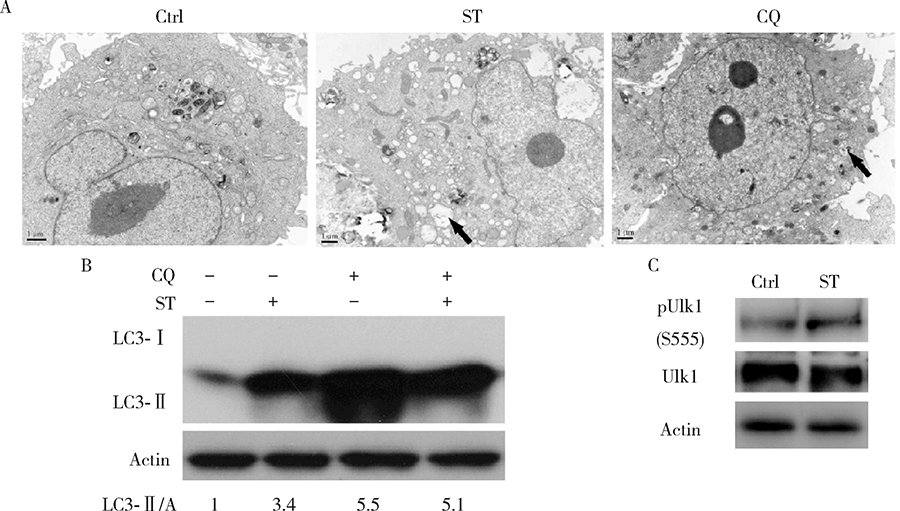 | 图2 氯喹和舒尼替尼增加786-O细胞内的自噬体Figure 2 CQ and ST increase the autophagosomes in 786-O cellsA, electron microscopy was performed in 786-O cells following treatment of sunitinib (ST, 8 μ mol/L) for 4 h, or chloroquine (CQ, 20 μ mol/L) for 2 h, and arrow indicates the autophagosome; B, 786-O cells were treated with ST (8 μ mol/L), or CQ (20 μ mol/L), or combination of them for 4 h; C, 786-O cells were treated with or without ST (8 μ mol/L) for 4 h. Then cell lysates were prepared and analyzed by immunoblotting using the indicated antibodies. Densitometry was performed for quantification, and the ratios of LC3-Ⅱ to actin (A) were presented below the blots. Ctrl, control. |
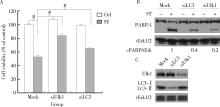
自噬与凋亡的关系密切, 自噬既可以促进也可以抑制凋亡的发生[20], 聚腺苷酸二磷酸核糖转移酶[poly (ADP-ribose) polymerase-1, PARP-1]在DNA修复和凋亡中发挥重要作用[21], 由于PARP-1可以被caspase-3切割, 所以其可以作为细胞发生caspase依赖的凋亡指标[22]。从图3A中可以看出, 舒尼替尼可以诱导细胞发生caspase依赖的凋亡。在氯喹存在的情况下, 舒尼替尼引起的PARP-1切割会明显的增加(图3A, 泳道2 vs. 泳道3)。而3-MA常常被用于抑制早期自噬[6], 其存在却减少了舒尼替尼引起的PARP-1切割, 说明抑制早期自噬和晚期自噬对舒尼替尼诱导的细胞凋亡作用不同(图3B, 泳道2 vs. 泳道5)。另外, 与3-MA不同, 雷帕霉素作为自噬诱导剂促进ST诱导的PARP-1切割, 即细胞凋亡(图3A, 泳道2 vs. 泳道6)。接下来, 实验进一步利用MTS检测了3-MA及雷帕霉素对舒尼替尼诱导的细胞活性丢失的影响。如图3C所示, 3-MA可以部分挽救舒尼替尼诱导的细胞活性丢失, 而雷帕霉素进一步降低了细胞活性。以上结果表明, 促进自噬增强了舒尼替尼诱导细胞凋亡和细胞活性丢失的能力。
 | 图3 自噬调节舒尼替尼诱导的细胞凋亡Figure 3 Autophagy regulates the ST-induced apoptosisA and B, 786-O cells were treated with indicated compounds (RAPA: 0.1 μ mol/L; 3-MA: 2 mmol/L) for 12 h, then cell lysates were prepared and analyzed by immunoblotting using the indicated antibodies, densitometry was performed for quantification, and the ratios of cleaved PARP-1 to tErk1/2 were presented below the blots; C, 786-O cells were treated with ST (0-4 μ mol/L) or together with Rapa (0.1 μ mol/L) or 3-MA (2 mmol/L), and cell viability was analyzed by MTS at the 24 h time point. ST, sunitinib; CQ, chloroquine. * P < 0.05, # P < 0.01 vs. control (Ctrl). |
为了进一步验证早期自噬水平对舒尼替尼诱导的细胞凋亡的影响, 786-O细胞中的LC3和Ulk1蛋白表达被siRNA技术特异性的敲除, 磷脂酰乙醇胺化的LC3, 即LC3-Ⅱ 是形成自噬体的关键蛋白, 而Ulk1在自噬的起始过程中发挥作用[8]。如图4A所示, 在MTS检测细胞活性实验中, 敲降Ulk1和LC3部分挽救了舒尼替尼诱导的细胞活性丢失。前面的结果显示舒尼替尼能够诱导细胞凋亡, 实验接下来进一步检测了敲降Ulk1和LC3是否会影响舒尼替尼诱导的细胞凋亡。免疫印迹结果显示, 在Ulk1和LC3敲降的786-O细胞中, 舒尼替尼诱导的PARP-1切割明显少于对照组细胞, 说明自噬基因的敲降在一定程度上抑制了细胞凋亡(图4B)。图4C显示, 敲降后相应的蛋白质表达明显降低, 说明实验系统是工作的。上述结果表明, 抑制早期自噬抑制了舒尼替尼的细胞毒作用和诱导凋亡的能力。
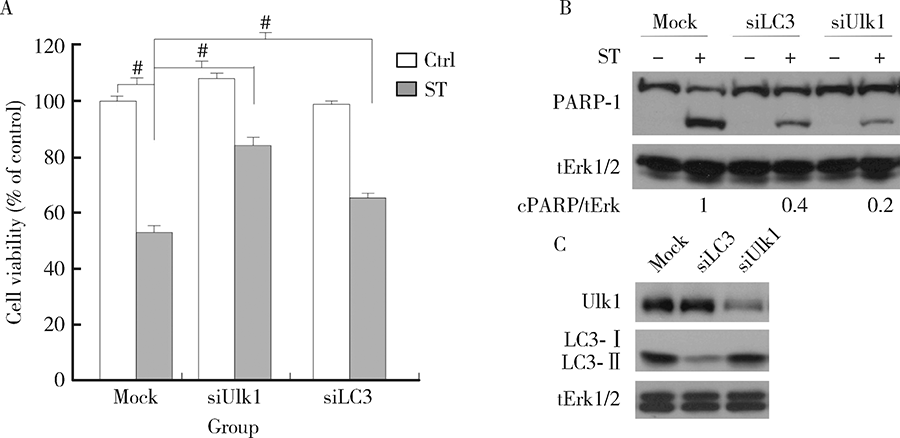 | 图4 干扰Ulk1或LC3挽救舒尼替尼诱导的细胞活性丢失Figure 4 Deprivation of either Ulk1 or LC3 rescue the ST-induced cell viability loss786-O cells were transfected with siRNA target Ulk1 or LC3 for 48 h. A, cell viability was analyzed by MTS assay after sunitinib(ST, 8 μ mol/L) treatment for 24 h in 786-O cells; B and C, cell lysates were analyzed by immunoblotting with the indicated antibodies following 12 h ST treatment in 786-O cells. Densitometry was performed for quantification, and the ratios of cleaved PARP-1 to tErk1/2 were presented below the blots. # P < 0.01 vs. control. |
随着研究不断深入, 越来越多学者认识到自噬可以作为肿瘤治疗的一个靶点。氯喹作为临床上治疗疟疾和类风湿性关节炎的药物, 因其能抑制自噬体和溶酶体的融合和引发晚期自噬而被用来作为潜在治疗肿瘤的药物进行临床前研究。氯喹是否可以用于肾癌治疗的研究工作, 目前还鲜有报道。本研究发现, 氯喹本身不仅能抑制肾癌细胞786-O的增殖, 同时还可以增加舒尼替尼的抑瘤活性。与抑制早期自噬不同, 氯喹促进了舒尼替尼诱导的细胞凋亡。
目前, 有关氯喹与肿瘤治疗的研究依然是靶向治疗的热点之一。在结肠癌中, 氯喹与化疗药物SN-38具有协同作用[23]。有研究发现, 氯喹可以通过影响肿瘤细胞的微环境引起乳腺癌细胞的凋亡从而抑制肿瘤[24]。对于肺癌, 发生胸腔积液就意味着预后不良。最近的研究发现, 氯喹可通过抑制蛋白激酶B(protein kinase B, PKB or AKT)抑制血管形成, 与对照组相比, 应用氯喹可以有效地减少胸腔积液的发生和动物肺部的瘤转移灶[25]。体外和体内的试验表明, 氯喹可以增加硼替咪唑(bortezomib)引起的黑色素细胞的凋亡[26]。上述研究表明, 氯喹有可能具有治疗恶性肿瘤的潜在价值而被用于临床。
自噬与凋亡的关系密切, 经过近几年的研究, 人们逐渐认识到, 自噬和凋亡总体存在着3种关系:一是自噬可以促进凋亡的发生, 如在自噬发生的过程中, 首先在细胞内出现双层膜结构的自噬体, 当细胞受到蛋白酶体抑制剂刺激时, 在自噬体形成的过程中将会激活caspase-8, caspase-8可以与FADD (fas-associated protein with death domain)、Atg5形成复合物, 进而导致细胞凋亡[27]; 二是自噬抑制凋亡的发生, 在细胞中, 自噬抑制凋亡发生最经典的机制是线粒体自噬[28]; 三是自噬和凋亡协同作用促进细胞死亡[29]。本研究发现, 无论是3-MA还是敲降自噬基因均可以抑制舒尼替尼诱导的细胞凋亡; 而氯喹的作用却恰恰相反, 它能促进前者引起的凋亡, 但具体的机制有待于进一步的研究来阐明。
舒尼替尼作为多种受体酪氨酸激酶抑制剂, 已经被美国FDA批准作为靶向治疗肾癌和其他肿瘤, 也被国内泌尿外科治疗指南批准为晚期肾癌一线治疗药物, 并可作为索拉菲尼等治疗失败的二线治疗药物。本研究表明, 氯喹不仅本身能抑制肾癌细胞的活性, 同时其与舒尼替尼具有协同抑制肿瘤细胞增殖的作用。深入进行这方面的研究, 将会为氯喹可能的临床应用打下理论基础。
(本文编辑:王 蕾)
The authors have declared that no competing interests exist.
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|