




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
基于光学结构识别技术的化学知识库构建
[吕传宇, 李明娜, 张亮仁, 刘振明△  ]
]
]
|
|


科学研究结果通常以文章或专利的形式发表, 阅读文献是获取医药研发信息最普遍的方式。药物化学家、医药情报分析师等在工作过程中需要查询大量的文献, 据统计, 出版化学类文章的学术期刊大约有10 000家[1], 每年在药物化学及生物化学领域公开的新化学结构超过20 000个[2]。将文献信息提取并存储为结构化的化学知识库, 通过数据库进行检索, 可以帮助科研工作者快速准确地获取情报, 提高工作效率, 同时, 化学知识库有利于期刊数据的长期保存以及科研成果的转化推广。
目前最权威的化学知识库是美国化学文摘服务社(Chemical Abstracts Service, CAS)建立的SciFin-der以及荷兰爱思唯尔公司建立的Reaxys。过去的100年间, CAS从全球招募成千上万的志愿者进行化学信息的挖掘, 时至今日, 超过500名科学家在CAS总部专门处理新披露的化学信息[3]。Reaxys将Beilstein、Patent和Gmelin三个数据库的内容整合为统一的资源, 其收录的数据上至18世纪, 包含400多万条文献[4], 由此可见, 建立高质量的化学知识库需要投入高昂的人力和时间成本。
传统的化学知识库是通过专业人员阅读文献、手动绘制化合物结构、人工注释信息的方法构建的, 这种方式确实需要消耗大量的人力、物力和财力。2014年开始, Journal of Medicinal Chemistry要求作者在投稿时提交含有化合物SMILES(Simplified molecular input line entry specification)和基本信息的表单, 这些文件可以直接导入数据库[5], 然而, 绝大多数文献只以非结构化数据的形式存在, 这些数据需要研究者主动去提取。为了节约时间、降低成本, 本研究借助光学结构识别、信息挖掘汇聚等技术, 提出了一种高效构建化学知识库的策略。
Intel Core i5 CPU 3.5GHz台式计算机有8 GB内存和1 024 MB显存, 使用Windows 10 Professional操作系统, 所使用的软件平台见表1。
| 表1 构建化学知识库所使用的工具 Table 1 Tools for construction of chemical information database |
文献中化合物的名称包括IUPAC名称、系统命名、通用名、商品名和CAS注册号等形式。本研究采用ChemAxon公司的化学信息挖掘工具J Chem Base 5.10.0[6]中的Naming模块将化合物名称转换为结构。目前, Naming支持将IUPAC名称、系统名称、通用名、药物的商品名以及部分CAS注册号转换成化学结构。使用Naming能够快速地将文档内的化合物名称转换为结构, 但这一工具有一定的局限性, 首先, 不规范的系统命名法无法识别; 其次, 通用名、商品名、CAS注册号的识别依赖于底层数据库的支持, 数据库中没有的名称无法转化为结构。
在期刊文献中, 存在大量图片形式的化合物结构, 图片结构本身并不能直接被计算机所识别, 必须通过一定手段将图片结构转换为可供编辑、检索的专业的计算机编码形式, 如SMILES、Mol、Mol2、InChI等[7]。过去, 人们只能依靠ChemDraw、MarvinSketch、BIOVIA Draw等化学绘图软件手动绘制化合物结构。随着光学文字识别技术的发展, 出现了一批可以从文档和图片中提取化学结构的软件工具, 如CLiDE、OSRA、Imago, 它们可以从图片中将化学结构识别出来, 并导出成为计算机可读的文件格式[8, 9, 10], 这一技术也被称为光学结构识别(optical structure recognition, OSR)。
本研究使用CLiDE Professional 5.12.1[11]来进行化合物结构提取, 将PDF按照一定规则重命名为系列编号, 然后导入CLiDE。CLiDE可以快速将PDF中的图片结构转换为计算机可识别的形式, 并存储到SDF(structure data file)文件中。SDF文件包含化合物结构、化合物所在页码、PDF编号等信息。
文献中的图片结构通常以两种形式存在:一种是具有明确结构的单体化合物, 单体结构可以使用CLiDE快速地提取保存; 另一种是族性化学结构, 即Markush结构。Markush结构是由一个新颖的母体基团和可变取代基组成[12], 由于取代基的不确定性和复杂性, 通常难以处理。本研究通过基团枚举的方式, 将Markush结构转变为单体结构, 根据基团数目和匹配方式的差异, 处理方式各不相同。
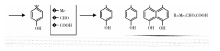
单一R基团Markush结构的处理见图1, CLiDE能够自动识别分子中的R基团, 并将不同取代基与R基团匹配, 生成所有结构。
 | 图1 单一R基团Markush结构的处理Figure 1 Processing of single R group Markush structure |
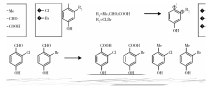
具有多个R基团且不同取代基可以任意组合的Markush结构的处理见图2, 使用CLiDE提取母核结构, 然后借助ChemDraw Ultra 14.0[13]中的QueryTools, 标记不同R基团的取代基, 进而枚举出所有的结构。假如一个母核有N个取代基, 每个取代基的种类分别是M1、M2…MN个, 通过这种方式可以快速得到M1× M2…MN个结构。
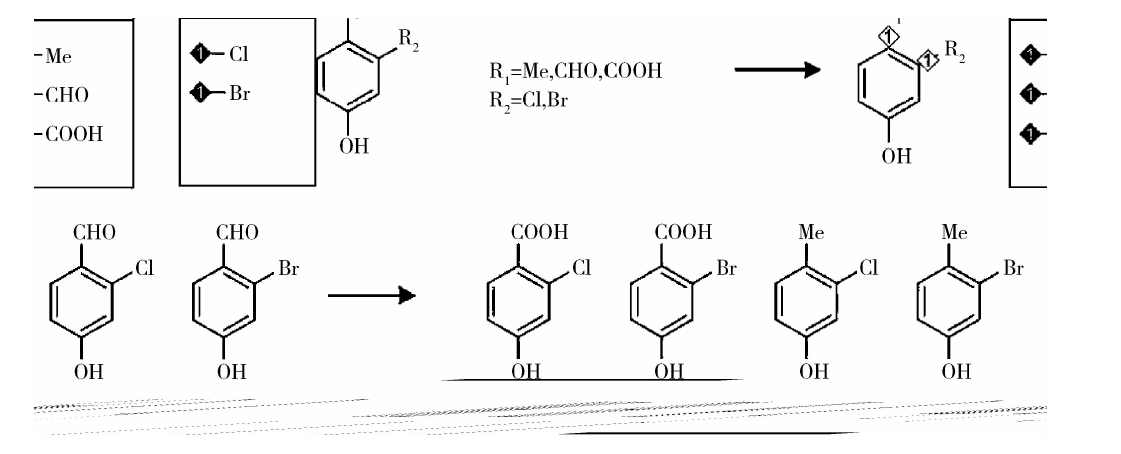 | 图2 任意组合的多R基团Markush结构的处理Figure 2 Processing of multi-R groups Markush structure whose substituents can be assembled arbitrarily |
具有多个R基团但取代基的排列组合固定的Markush结构的处理见图3, 目前的OSR工具很难识别表格中R基团的逻辑对应关系, 本研究使用CLiDE提取母核结构, 然后在ChemDraw中根据编号手动匹配不同的取代基。
 | 图3 固定组合的多R基团Markush结构的处理Figure 3 Processing of multi-R groups Markush structure whose substituents assembly is specified Cmpd, compound identity. |
为了保持数据的完整性和可追溯性, 本研究对期刊发表的文章的题录进行了提取, 所用工具为EndNote X8[14]。将统一编号的PDF文档导入EndNote, 更新文献信息, 自定义Output Style, 保留文章标题、作者、作者单位、期刊名称、发表时间以及文献的DOI号, 然后利用EndNote的导出功能将题录信息存储为便于处理的文本形式, 最后得到含有PDF编号和题录信息的TXT文档。
1.4.1 信息挖掘 生物医学领域已经进入大数据时代, 与日俱增的海量科学文献迫切需要使用计算机技术进行统一的存储与管理。为了将文献内容转化成结构化的数据库, 研究者将机器学习应用到化学信息挖掘中来。本研究采用ChemDataExtractor[15]进行信息挖掘工作, ChemDataExtractor是剑桥大学卡文迪什(Cavendish)实验室开发的一款可以自动从科学文献中提取出化学信息的工具。文档处理器读取PDF内容并拆分, 使用自然语言处理流程对片段化的内容进行语义分析, 经过监督学习和非监督学习的训练, 识别不同属性及参数, 相关的数据被合并成记录。ChemDataExtractor可以提取化合物的熔沸点、反应收率、磁共振数据、紫外-可见光谱、红外光谱等信息。
1.4.2 人工注释 人工阅读文献并提取关键信息是构建数据库传统的方式, 由于PDF文档内的数据并非完全符合计算机处理的规范, 错误和遗漏不可能完全消除, 许多信息仍需要人工提取, 这能够保证数据的完整性和准确性。
本研究采用Accelrys公司的计算模拟和信息处理平台Pipeline Pilot 7.5.2[16]进行属性预测。将从文献中提取的化合物SDF文件导入Pipeline Pilot并生成Canonical SMILES。通过结构模块计算化合物的相对分子质量、氢键给体数、氢键受体数、可旋转键数等参数。通过物理化学性质模块计算化合物的脂水分配系数、溶解度、pKa、极性表面积。通过ADMET模块计算化合物在生物体内的吸收、分布、代谢、排泄和毒性特征, 包含的模型有:人体肠内吸收预测模型、水溶性预测模型、血脑障碍渗透性预测模型、血浆蛋白结合率预测模型、细胞色素P450 2D6抑制率预测模型和肝毒性预测模型。
ChEMBL数据库是欧洲生物信息研究所开发的一个在线的免费数据库, 它通过从大量文献中收集各种靶点及化合物的生物活性数据, 为药物化学家们提供了一个非常便利的查询靶点或化合物的生物活性数据平台[17]。
最新的ChEMBL_23数据库包含靶点11 538个, 活性小分子1 735 442个, 活性信息14 675 320条[18]。将ChEMBL开源数据下载至本地, 并进行标准化处理。经过整理, 得到具有明确结构的小分子1 069 420个, 活性记录7 871 105条, 包含靶点名称以及IC50、Ki等实验数据。使用Pipeline Pilot搭建流程, 将从文献中提取出的化合物的Canonical SMILES作为关键词在ChEMBL本地数据库中检索, 得到与之匹配的活性信息。
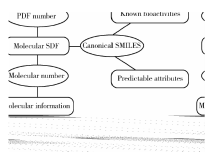
经过以上5步操作, 得到5份独立的元数据文件:化合物SDF文件、题录信息、文献内化合物信息、已知生物活性以及可预测属性。借助PDF编号、化合物编号、Canonical SMILES 3个节点进行数据关联(图4)。
 | 图4 数据关联示意图Figure 4 Schematic diagram of data association |
CLiDE从PDF文档中提取的化合物SDF文件包含化合物结构、化合物编号及PDF编号。通过PDF编号与EndNote导出的题录信息关联, 通过化合物编号与ChemDataExtractor抓取的文献内化合物信息关联。SDF文件导入Pipeline Pilot后生成的Canonical SMILES作为数据库主键, 通过Canonical SMILES分别与从ChEMBL获得的已知生物活性以及Pipeline Pilot预测的化合物属性关联。
搭建了一套完整的化学知识库构建流程(图5), 按照此流程对科学文献进行处理, 可以较快速地得到含有结构信息和属性信息的化学知识库。
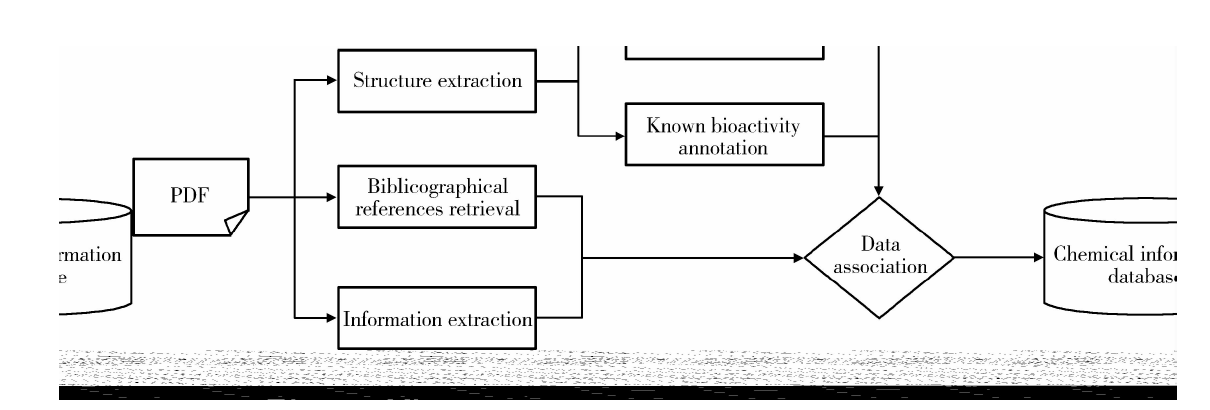 | 图5 化学知识库构建流程Figure 5 Schematic map of chemical information database construction |
本研究对2015年1月至2016年6月发表在期刊Marine Drugs上的174篇研究性文章和25篇综述进行信息提取, 初步建立起北京大学海洋天然产物库PKU-MNPD。目前, 数据库包含3 262个结构明确的化合物, 数据记录19 821条, 部分数据样本见表2~4。
| 表2 北京大学海洋天然产物库数据样本(结构& 题录信息) Table 2 Data sample of Peking University marine natural product database (structures & bibliographical references) |
| 表3 北京大学海洋天然产物库数据样本(可预测属性) Table 3 Data sample of Peking University marine natural product database (predictable attributes) |
| 表4 北京大学海洋天然产物库数据样本(生物活性) Table 4 Data sample of Peking University marine natural product database (bioactivities) |
根据名称在大型化学数据库如PubChem[19]检索, 或者依照图片手工绘制, 是从科学文献提取化学结构最常用的方式。本研究采用Naming技术和OSR工具, 极大地缩短了提取结构所使用的时间, 对于结构复杂的大分子天然产物尤为适用。采用机器学习对文献内容进行语义分析, 快速定位关键信息, 辅以人工注释, 保证了数据库构建的高效性和准确性。
跨数据库实现数据交流, 是扩充数据库容量的有效方式。本研究以化合物的Canonical SMILES为主键, 关联ChEMBL数据库中的已知生物活性信息, 同时采用计算模拟平台Pipeline Pilot预测化合物的物理化学性质、ADMET等参数, 使数据库信息更全面。
化学知识库构建的目的是方便科研人员快速查找相关信息, 本研究建立的化学知识库支持结构搜索和文本搜索, 具有检索方式多样化的特点。结构搜索支持精确结构查询、子结构查询以及相似性匹配查询。文本搜索支持通过化合物分子式、SMILES、物理化学性质、生物活性等字段单途径或任意组合进行检索。
综上所述, 本研究提出的化学知识库构建策略能够帮助医药研发人员高效地建立数据库, 同时, 基于原始文献构建的化学知识库内容准确、全面, 检索方式多样, 可以帮助科研人员精准地获取信息。
The authors have declared that no competing interests exist.
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|