Journal of Peking University (Health Sciences) ›› 2026, Vol. 58 ›› Issue (2): 327-331. doi: 10.19723/j.issn.1671-167X.2026.02.016
Previous Articles Next Articles
Genetic variants analysis of 17 female patients with idiopathic hypogonadotropic hypogonadism
Qiqi CHEN1, Haining WANG2, Ye LIU2, Xu ZHI1,*( )
)
- 1. Department of Obstetrics and Gynecology, Center for Reproductive Medicine, Peking University Third Hospital; State Key Laboratory of Female Fertility Promotion; National Clinical Research Center for Obstetrics and Gynecology (Peking University Third Hospital); Key Laboratory of Assisted Reproduction (Peking University), Ministry of Education; Beijing Key Laboratory of Reproductive Endocrinology and Assisted Reproductive Technology, Beijing 100191, China
2. Department of Endocrinology and Metabolism, Peking University Third Hospital, Beijing 100191, China
CLC Number:
- R394.8

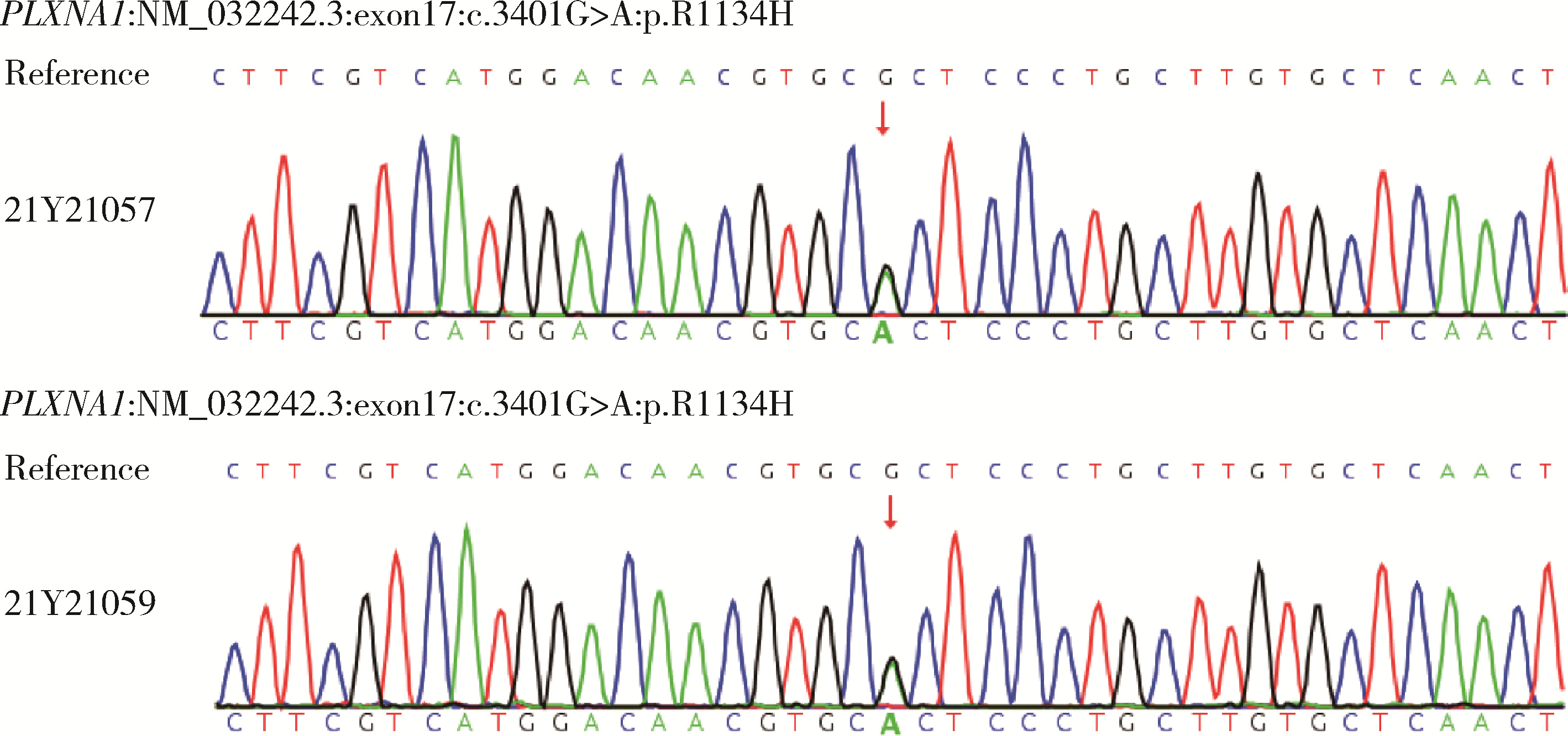
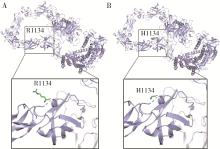
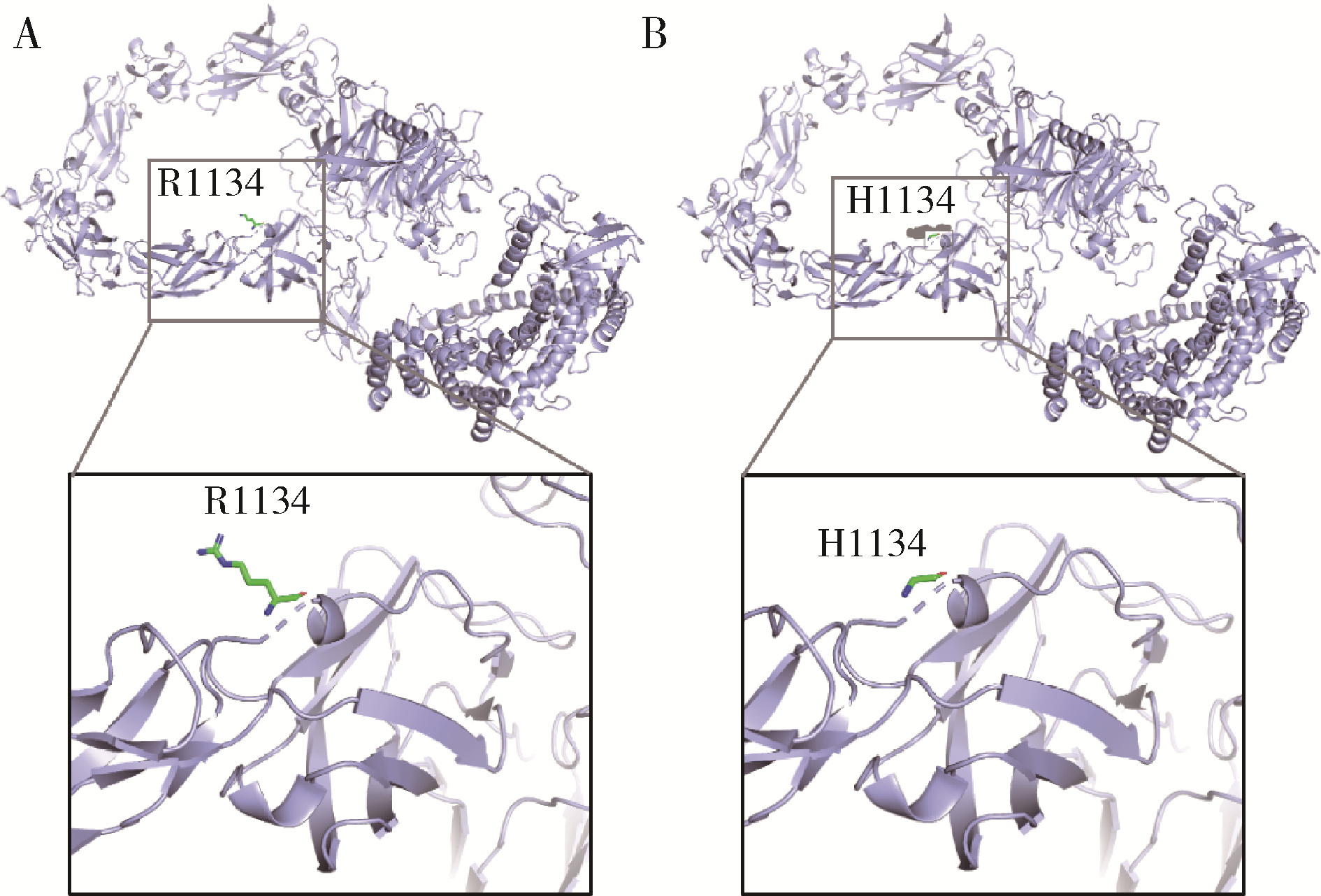
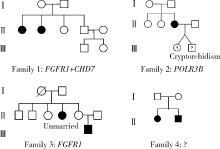
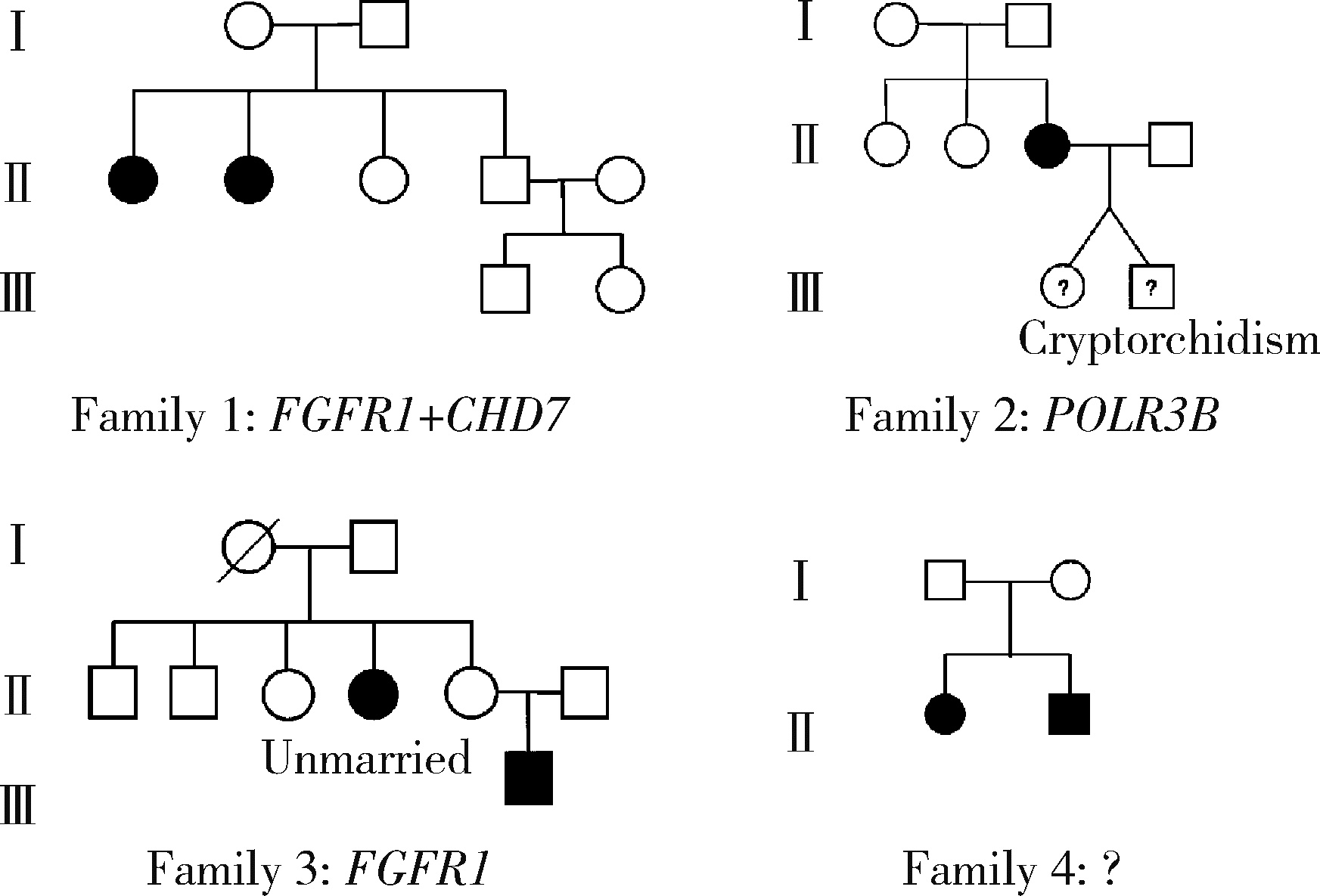
| 1 |
doi: 10.1186/1750-1172-6-41 |
| 2 |
doi: 10.3389/fendo.2022.965074 |
| 3 |
doi: 10.1111/cen.14822 |
| 4 |
doi: 10.1093/hmg/ddx080 |
| 5 |
doi: 10.1242/dev.176461 |
| 6 |
汪保安, 马晓莉, 邹效漫, 等. 女性特发性低促性腺激素性性腺功能减退症的临床评价及KAL1和FGFR1基因突变分析[J]. 军医进修学院学报, 2011, 32 (10): 1017- 1019.
|
| 7 |
曾旺, 李家大, 王新颖, 等. 中国先天性低促性腺激素性性腺功能减退症患者ANOS1的突变(英文)[J]. 中南大学学报(医学版), 2022, 47 (7): 847- 857.
|
| 8 |
张丽琼, 张红艳, 李明泸, 等. 低促性腺激素性性腺功能减退症患者KAL-1基因突变分析[J]. 云南医药, 2010, 31 (1): 27- 30.
|
| 9 |
解一丹, 郑瑞芝, 韩宾宾, 等. 22例低促性腺激素性性腺功能减退症患者CHD7基因变异分析[J]. 中华医学遗传学杂志, 2022, 39 (6): 571- 575.
|
| 10 |
doi: 10.1002/mgg3.1816 |
| 11 |
doi: 10.1111/cge.13482 |
| 12 |
|
|
||