
系统性红斑狼疮(systemic lupus erythematosus,SLE)是一种自身免疫介导的全身性疾病,可累及中枢和周围神经系统。狼疮相关周围神经系统受累的研究相对较少见,根据美国风湿病学会(American College of Rheumatology,ACR)的定义,包括急性炎性脱髓鞘性多神经根神经病(acute inflammatory demyelinating polyradiculoneuropathy,AIDP)即格林-巴利综合征(Guillain-Barré syndrome,GBS)、自主神经紊乱、单神经病变、重症肌无力、颅神经病变、神经丛病变和多发性神经病共七类,其他类型如慢性炎性脱髓鞘性多神经根神经病(chronic inflammatory demyelinating polyradiculoneuropathy,CIDP)目前尚未被正式纳入。
近年发现部分被诊断为CIDP的患者存在靶向郎飞结及结旁区的特异性抗体,此类患者的临床表现、免疫和病理特征及治疗反应均与传统CIDP存在差异。随着研究的深入,学界逐渐意识到此类疾病并非CIDP的一个变异亚型,而是一类独立的免疫性神经病,因此在2021年7月,欧洲神经病学会(European Academy of Neurology,EAN)联合周围神经协会(Peripheral Nerve Society,PNS)首次正式提出自身免疫性郎飞结病(autoimmune nodopathy,AN)这一定义[1]。目前,SLE患者合并AN的相关报道非常罕见[2]。本文报道1例SLE合并抗接触蛋白1(contactin-1,CNTN1)阳性AN患者,并结合文献探讨其发病机制、临床特征、治疗及预后,以期为相关疾病的早期识别与精准管理提供参考。
1 资料与方法
1.1 一般资料
收集1例于2023年3月在北京大学人民医院风湿免疫科诊断为SLE合并AN患者的病例资料,并长期随诊,记录患者的性别、年龄、就诊原因、化验检查及病理诊断结果。
1.2 研究方法
采用回顾性研究方法对病例的临床特点和随诊数据进行分析,并对相关文献进行复习。
2 结果
2.1 临床资料
患者女性,48岁,2021年10月无诱因出现双侧足趾对称性麻木,逐渐进展累及双手十指指尖、唇周及舌尖,伴四肢远端无力、双足“踩棉花感”、双手精细动作笨拙及轻度震颤。于当地医院检查尿常规示蛋白+、红细胞35/高倍视野。腰椎穿刺脑脊液无色透明,细胞总数25×106/L(淋巴细胞78%、单核细胞16%)、总蛋白2 092.1 mg/L。肌电图提示上下肢运动神经传导速度减慢、波幅减低、潜伏期延长,右腓总神经、双正中神经可见传导阻滞;上下肢感觉神经传导速度减慢、波幅减低、潜伏期延长;双下肢F波未引出,上肢F波潜伏期延长;双下肢H反射缺失;针极肌电图未见明显异常,初步诊断为“周围神经病”,予静脉注射人免疫球蛋白(intravenous immunoglobulin,IVIG)22.5 g×4 d及营养神经治疗,疗效欠佳。症状持续进展,出现近端肢体无力及站立不稳。患者转诊至上级医院,查血清抗单唾液酸四己糖神经节苷脂4(monosialotetrahexosylganglioside 4,GM4)抗体免疫球蛋白G(immunoglobulin G,IgG)和抗双唾液酸神经节苷脂1(disialosylganglioside 1,GD1)抗体IgM阳性,红细胞沉降率65 mm/h。复查针极肌电图提示双下肢远端肌肉神经源性损害,静息状态下可见纤颤电位及正锐波,诊断为“CIDP”。予甲泼尼龙500 mg/d×4 d、240 mg/d×6 d、120 mg/d×4 d、80 mg/d×3 d静脉滴注,序贯口服泼尼松60 mg隔日1次,每周减量10 mg隔日1次至停药。症状无明显缓解,并缓慢加重至行走需搀扶、双手握力显著下降。2022年7月,患者出现双下肢水肿,复查血常规白细胞3×109/L,尿常规蛋白+++、潜血+++,尿蛋白/肌酐4.5 g/g,血清白蛋白23.84 g/L,总胆固醇6.05 mmol/L、低密度脂蛋白4.05 mmol/L、甘油三酯2.36 mmol/L。血清郎飞结抗体谱:抗CNTN1抗体1 ∶ 320,IgG2 1 ∶ 10、IgG4 1 ∶ 100。腰椎MRI示双侧腰骶神经束增粗,腓总神经神经束水肿,臂丛神经弥漫性增粗。诊断为“免疫介导性周围神经病、肾病综合征”,予利妥昔单抗600 mg静脉滴注1次,地塞米松15 mg/d静脉滴注5 d,后序贯泼尼松60 mg/d,每周递减5%直至停药。经以上治疗患者肢体无力,口周、舌尖麻木均较前好转,可缓慢行走,复查白细胞升至正常,但双下肢水肿逐渐加重。2023年1月,患者脱发明显,无颜面红斑、光过敏、关节肿痛、口眼干、唾液腺肿大或雷诺现象等。2023年3月于外院复查尿蛋白/肌酐7.05 g/g、血清白蛋白19.87 g/L。完善免疫学检测:抗核抗体(antinuclear antibody,ANA)1 ∶ 1 000胞浆颗粒型,抗SSA、Ro52、双链DNA (double-strand DNA,dsDNA) 抗体阳性,抗SSB抗体弱阳性。血清与脑脊液抗CNTN1、GM4及GD1抗体均阴性,脑脊液细胞总数1×106/L、总蛋白185 mg/L。神经超声提示左侧正中神经上臂近段较对侧略粗、右侧正中神经肘部水平较对侧略粗,左侧尺神经上臂近段较对侧略粗,左侧臂丛神经C7、C8较对侧稍粗,C7内瘢痕影像,左侧腓总神经腓骨颈水平较对侧增粗,双侧胫神经远段增粗。考虑系统性红斑狼疮,为进一步诊治,于2023年3月收入北京大学人民医院住院治疗。
2.2 入院后评估
入院查体:跨阈步态,四肢肌力Ⅴ级,双足前掌浅感觉及膝以下痛觉减退,双下肢腱反射减弱,Babinski征阴性,共济运动基本正常,双侧胫前可凹性水肿。辅助检查:血常规未见异常,尿液蛋白电泳见许多相对分子质量68 000蛋白、微量相对分子质量160 000蛋白,尿红细胞形态见草莓状56%、面包圈24%、皱缩20%。ANA 1 ∶ 160胞浆颗粒型,抗SSA、抗Ro52抗体+++,抗dsDNA抗体137 IU/mL,抗磷脂酶A2受体(phospholipase A2 receptor,PLA2R)抗体阴性,C4 0.37 g/L,外周血CD19+ B细胞23 /μL,血尿免疫固定电泳、Hu-Yo-Ri未见异常。眼科检查除外干眼症,唾液腺显像未见明显异常。肾穿刺活检光镜提示肾小球系膜细胞及基质轻度弥漫增生,局灶节段性中度加重,基底膜增厚,伴多数节段性钉突形成,上皮下、系膜区嗜复红蛋白沉积;肾小管上皮细胞空泡及颗粒变性,灶状萎缩;肾间质灶状淋巴细胞及单核细胞浸润伴纤维化;小动脉管壁增厚(图 1)。免疫荧光可见3个肾小球,IgA++,IgG++,IgM++,C1q++,C3++,FRA-,PLA2R-,沿毛细血管壁及系膜区呈颗粒及团块样沉积。IgG亚型示IgG1++,IgG2+,IgG3++,IgG4+++,沿毛细血管壁及系膜区呈颗粒及团块样沉积(图 2)。电子显微镜提示肾小球系膜细胞和基质增生,基底膜弥漫不规则增厚,上皮下、基底膜内、内皮下和系膜区电子致密物沉积,上皮细胞足突弥漫融合,综上,符合膜性肾病(图 3)。
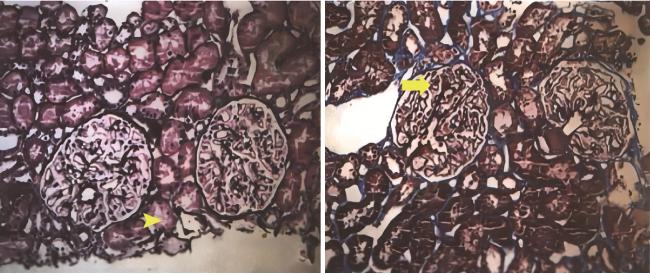
图1 肾活检光学显微镜下表现Figure 1 The light microscopic findings of renal needle biopsy Light microscopy showed glomerular mesangial hyperplasia (yellow→) and lymphocytic and monocytic infiltration in the renal interstitium with fibrosis (yellow▶). |
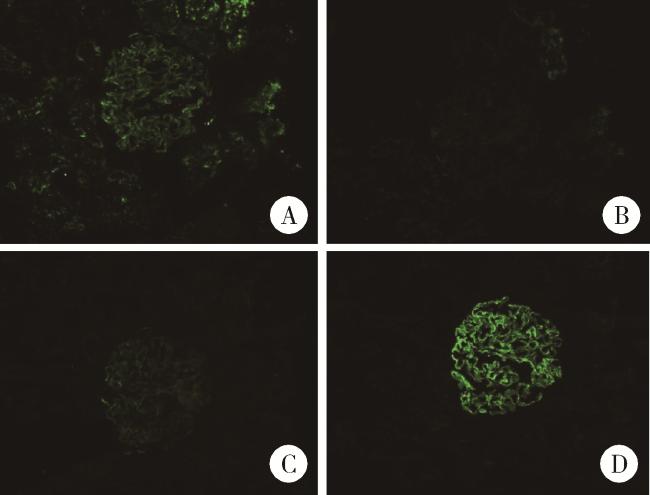
图2 肾活检免疫荧光表现Figure 2 The immunofluorescen findings of renal needle biopsy Immunofluorescence IgG subclasses showed IgG1++ (A), IgG2+ (B), IgG3++ (C), IgG4+++ (D). |
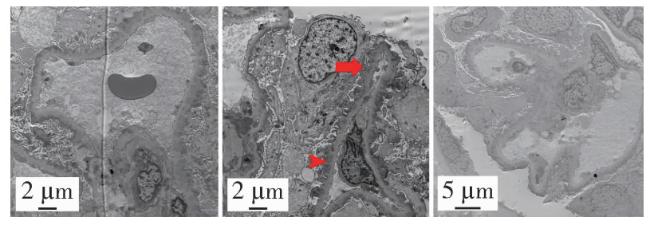
2.3 诊治及随访
经多科室会诊并讨论,诊断为系统性红斑狼疮、抗CNTN1抗体阳性自身免疫性郎飞结病、狼疮性肾炎(Ⅴ型)。患者经此前激素及利妥昔单抗治疗后神经系统症状较前改善,脑脊液细胞数及蛋白水平明显降低,抗CNTN1、GM4及GD1抗体转为阴性,提示治疗存在一定疗效;但其肾受累持续、复查仍存在血清学活动,推测可能与激素快速减停及利妥昔单抗单次治疗后未进行序贯治疗相关。加用甲泼尼龙40 mg/d静脉滴注、羟氯喹0.2 g每日2次口服、利妥昔单抗500 mg/周、连续4周,并辅以低分子肝素序贯华法林抗凝(随诊期间患者尿蛋白逐渐减少、血清白蛋白上升到大于25 g/L并维持稳定后停药,疗程共6个月),维生素B1、甲钴胺片营养神经,以及碳酸钙、骨化三醇补钙等综合治疗。
经以上治疗后,患者脱发好转,肢体无力逐渐减轻、无震颤,可不借助辅助行走。化验尿蛋白/肌酐逐渐减少,抗dsDNA、抗CNTN1、GM4及GD1抗体持续阴性。出院后患者每3~6个月随诊1次,激素逐渐减量至泼尼松7.5 mg/d、每6个月序贯利妥昔单抗500 mg 1次。间断监测外周血CD19+ B细胞,利妥昔用药3个月后可降为0~1 /μL、6个月后回升为10~17 /μL。自北京大学人民医院就诊至末次随诊(2025年5月)已2年余,患者尿蛋白/肌酐降至0.19 g/g。
3 讨论
3.1 SLE与CIDP
SLE合并周围神经病变的发病率为5%~27%,发病机制可能涉及免疫复合物沉积、血管炎、特异性自身抗体介导、细胞毒性T细胞激活及代谢紊乱等多个方面。合并周围神经系统受累的SLE患者多呈现更高的疾病活动度,生活质量显著下降。然而,尽管CIDP在SLE患者中并不罕见[3],目前ACR对于SLE相关周围神经病变的分类中尚未将其纳入,在临床识别与管理上造成了一定困难。
3.2 CIDP与AN
进一步研究发现,与典型CIDP不同,此类患者具有独特的临床、免疫病理及组织学特征。临床表现方面,除CIDP典型的对称性四肢无力、感觉异常外,患者常合并感觉性共济失调、震颤、颅神经受累等非典型症状。与CIDP类似,AN患者的脑脊液检查多呈现“蛋白-细胞分离”现象,但程度往往更显著。与CIDP不同的是,AN患者的神经活检缺乏常见的巨噬细胞介导脱髓鞘反应及“洋葱球”样结构,取而代之的是结旁区连接髓鞘与轴突的隔膜样结构消失,导致郎飞结区扩大和结旁区脱髓鞘改变[7]。更为关键的是,此类患者对标准治疗方案中的静脉注射IVIG反应不佳,而对B细胞清除治疗如利妥昔单抗更为敏感,从另一侧面提示其发病机制与传统CIDP存在本质区别。在这一系列临床、免疫及病理特征,以及治疗反应和预后研究的推动下,学界逐渐意识到此类疾病并非CIDP的一个变异亚型,而代表一类独立的免疫性神经病,最终在2021年7月,EAN/PNS首次正式提出自身免疫性郎飞结病这一术语,将其作为一个独立于CIDP的由郎飞结/结旁区自身抗体介导的新的疾病谱系,并推荐在诊断和治疗策略中予以区别对待[2]。
3.3 抗CNTN1抗体阳性AN的特征与机制
致病抗体的差异导致自身免疫性郎飞结病呈现出明显的异质性。在这一复杂的疾病谱系中,CNTN1作为结旁区的一种轴突蛋白,属于免疫球蛋白超家族的细胞黏附分子,它与Caspr1形成异源二聚体,并与Schwann细胞侧的NF155结合,构成隔膜样连接,对维持郎飞结区离子通道聚集具有关键作用[8]。CNTN1是维持跳跃式神经传导的基础,在基因敲除小鼠模型中,CNTN1的缺失可导致神经传导速度的下降。抗CNTN1抗体通过攻击郎飞结及结旁区,破坏CNTN1/Caspr1/ NF155复合体结构,进而导致有髓神经纤维脱髓鞘及传导功能障碍。
抗CNTN1抗体以IgG4亚型为主,通常在慢性抗原刺激下产生,具有对Fc受体亲和力低、不参与经典补体途径的激活等特征,这一特性解释了此类患者对IVIG治疗的反应普遍较差[13]。但也有研究发现,在IgG4阳性的患者中,尤其在疾病早期,IgG1、IgG2、IgG3等亚型可能同时存在,能够诱导补体沉积和激活,因此在疾病早期阶段,部分患者仍可能对IVIG治疗有效[14]。目前,糖皮质激素仍然是抗CNTN1抗体阳性AN的一线治疗手段,大多数患者可获得一定缓解。对于一线治疗反应不佳者,靶向CD20的利妥昔单抗等B细胞耗竭疗法具有良好疗效,这与此类药物在其他类型IgG4介导疾病中的应用效果类似,目前,抗CNTN1抗体阳性AN中利妥昔单抗的用药剂量及疗程尚未形成统一推荐方案,用药方案呈现多样性,包括单次静脉滴注1 000 mg,隔周分次静脉滴注2剂1 000 mg,还有在初始治疗后每6个月序贯静脉滴注1次利妥昔以维持疗效。值得注意的是,有临床观察发现,未接受规律序贯治疗的患者,在长期随访中更易出现疾病复发,而复发患者再次接受利妥昔单抗治疗后,多数可重新获得症状缓解[9]。此外,也有使用吗替麦考酚酯等免疫抑制剂治疗的个案报道[15]。血浆置换也可作为桥接治疗手段,用于快速清除循环抗体,降低疾病负荷[16]。
3.4 SLE与抗CNTN1抗体阳性的AN
目前SLE患者合并AN的相关报道非常罕见,截止本文投稿前查阅文献仅见两例相关报道。Tang等[2]报道了首例诊断SLE合并抗CNTN1抗体阳性的AN患者,除周围神经及肾受累表现外,血清学检测到ANA及抗dsDNA阳性,患者临床诊断系统性红斑狼疮及狼疮性肾炎,但未行肾活检,是否存在典型的狼疮临床特征也并未详细阐明。近期,Shida等[17]报道了1例抗CNTN1抗体阳性,同时出现周围神经及肾受累,血清学检测提示ANA、抗dsDNA及抗心磷脂抗体阳性的患者,肾病理符合膜性肾病,免疫荧光检查呈现“满堂亮”模式,即沿肾小球基底膜和系膜区可见IgG、IgA、IgM、C3、C4及C1q均呈阳性,考虑符合狼疮性肾炎(Ⅴ型)的诊断,但作者同时指出,此例患者未表现出SLE的任何特征性临床症状,无低补体血症,也未出现狼疮性肾炎的特异性病理改变, 如电子致密物中的指纹结构或肾小球细胞内的管状网状包涵体。因此,SLE与抗CNTN1抗体阳性AN的共病关系目前仍不明确。
本例患者病程中存在脱发、白细胞减少、周围神经病变及肾受累表现,ANA、抗dsDNA、CNTN1、GM4及GD1抗体阳性,补体减低,经激素及利妥昔治疗后脱发、周围神经病变明显缓解,白细胞恢复正常,尿蛋白明显减少,复查抗dsDNA、CNTN1、GM4及GD1抗体均转为阴性。从整体临床特征及治疗反应来看,最终诊断为SLE合并自身免疫性郎飞结病及膜性肾病(Ⅴ型狼疮性肾炎)。笔者推测,在免疫病理机制方面,SLE本身是一种以B细胞功能亢进、多克隆自身抗体产生为特征的自身免疫病,而CNTN1作为郎飞结区的关键黏附分子,其抗体的产生可能与SLE患者免疫耐受破坏后的“表位扩展”有关[11, 18],初始免疫应答针对核抗原(如dsDNA),后续交叉反应或分子模拟机制诱导机体对神经组织及肾组织抗原(如CNTN1)产生自身抗体[19]。本文患者同时存在抗GM4及GD2抗体,这种“多抗体共存”模式也更加支持SLE继发性神经病变,而非单一抗原靶向的原发性自身免疫性神经病。GM4作为髓鞘糖脂成分,其抗体可能直接破坏髓鞘结构稳定性,加剧脱髓鞘;抗GD1抗体则可能影响轴突膜的离子通道功能,导致轴突传导异常或轴索损伤。此外,SLE患者的免疫复合物沉积、补体激活及T细胞异常活化,可能进一步加重郎飞结区及肾脏的炎症损伤,形成“系统性免疫紊乱-神经-肾脏局部损伤”的恶性循环。需要说明的是,由于北京大学人民医院肾脏病理检测平台暂不具备检测肾组织CNTN1表达的能力,未能直接证实肾组织中CNTN1的存在及抗CNTN1抗体在肾组织的沉积情况。肾组织层面直接证据的缺失,是本研究的局限性所在。治疗方案选择上,鉴于SLE合并抗CNTN1抗体阳性AN的临床报道罕见,尚无统一用药标准,本例患者的利妥昔单抗治疗方案综合借鉴了抗CNTN1抗体阳性AN[8]、SLE[20]及膜性肾病[21]的临床常规用法,采用每周375 mg/m2体表面积静脉滴注,连续治疗4周;待病情稳定后,每6个月序贯静脉滴注1次以维持疗效。经激素联合利妥昔单抗治疗后,患者神经系统症状明显缓解,肾脏受累指标显著降低,血清抗dsDNA、抗CNTN1等抗体持续转阴,提示治疗方案有效。本例患者随诊期间,对患者外周血CD19+ B细胞水平进行了定期监测,经利妥昔单抗治疗后3个月左右复查提示外周血CD19+ B细胞可达到基本耗竭, 6个月后复查恢复。CD19+ B细胞具有检测便捷和可重复性强的优势,能快速量化利妥昔单抗介导的B细胞耗竭程度,为评估治疗起效与否、确定序贯治疗时机及预测疗效预后提供了直观依据;但需注意,外周血CD19+ B细胞水平无法完全反映肾、神经组织等靶器官内的B细胞耗竭状态,部分患者即使外周血B细胞已降至目标范围,组织内仍可能残留活化B细胞并驱动疾病活动,因此需结合抗dsDNA抗体、补体水平、尿蛋白定量等疾病活动指标综合判断病情,避免单纯依赖外周血指标导致的评估偏差。
综上所述,本课题组建议扩大SLE相关周围神经病变的定义,在合并对称性四肢无力及感觉异常,尤其在伴有感觉性共济失调、震颤及颅神经受累等症状,伴发肾病综合征表现,且对常规CIDP治疗反应不佳的患者,应考虑自身免疫性郎飞结病尤其是抗CNTN1抗体相关自身免疫性郎飞结病的可能。早期检测结旁区相关抗体,有助于明确诊断并选择针对性治疗,如B细胞耗竭疗法,从而促进神经功能恢复及改善患者长期预后。

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}