
VEXAS综合征是一种罕见、获得性、迟发性、进行性自身炎症性疾病,包括全身炎症表现及血液系统表现。全身炎症主要表现为发热、肺部受累(包括肺浸润、胸腔积液、中等大小支气管动脉血管炎、中性粒细胞肺泡炎等)、皮肤血管表现(包括嗜中性皮肤病、耳鼻软骨炎、复发性多软骨炎、结节性多动脉炎和巨细胞动脉炎等);血液系统疾病多表现为大细胞性贫血、血小板减少症、血栓栓塞性疾病、骨髓增生异常综合征等[1]。鉴于VEXAS综合征发病特点,目前尚无权威分类标准,易出现漏诊、误诊,多数患者依赖持续性大剂量糖皮质激素和靶向治疗以实现疾病缓解,但VEXAS综合征复发率及病死率较高。本文报道1例VEXAS综合征病例,模拟复发性多软骨炎、嗜中性皮肤病表现,同时出现血液系统受累,应用糖皮质激素联合磷酸芦可替尼、丙种球蛋白治疗,病情逐渐好转。现将本例患者诊治经过报告如下,以供临床医生参考。
1 病例资料
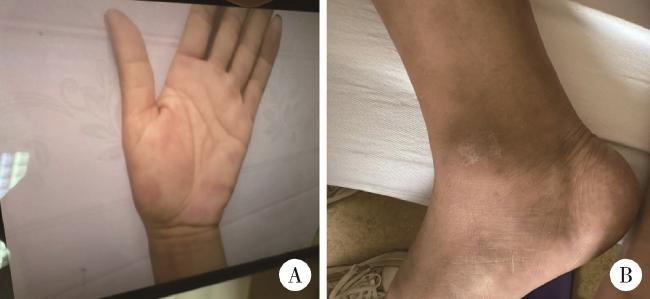
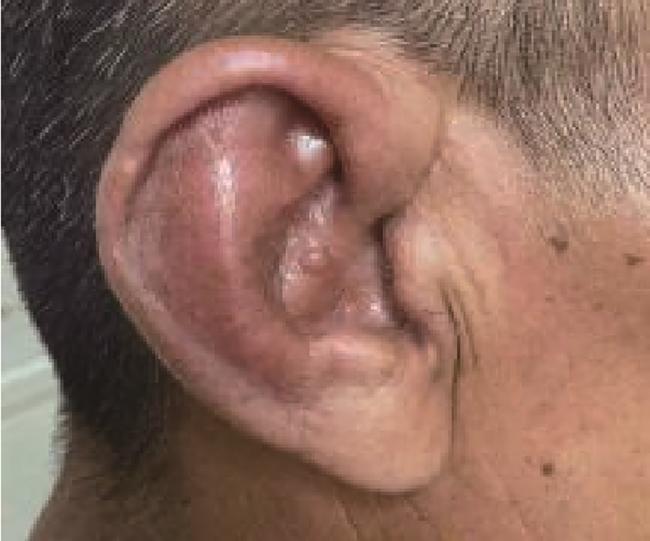
患者男性,53岁,主因“间断皮下结节4月,双侧耳廓肿痛3周”于2024年8月15日入北京大学人民医院风湿免疫科住院治疗。患者2024年4月以全身多发皮下结节伴疼痛起病,分布于四肢、双手掌部,伴四肢局部皮肤红肿(图 1),劳累或长时间行走后症状明显,伴轻微口干,无眼干、反复口腔溃疡、外阴溃疡,无发热,就诊于当地医院,查血常规:白细胞2.97×109 /L、血红蛋白96 g/L、血小板154×109 /L,红细胞沉降率(erythrocyte sedimentation rate,ESR)87 mm/h,C反应蛋白(C-reactive protein,CRP)96.47 mg/L,ANA及抗ENA抗体谱阴性,抗人球蛋白试验阴性,男性肿瘤标志物未见异常,血/尿免疫固定电泳阴性,予以利可君对症治疗,血细胞升高不明显,皮下结节仍反复。2024年6月于外院皮肤科行右前臂皮肤红肿部活检,病理示真皮中血管周围淋巴细胞浸润,局灶大量中性粒细胞浸润,呈嗜中性皮肤病改变,予以硫酸羟氯喹片0.2 g每日2次、雷公藤多苷片20 mg每日3次治疗,口服半月自觉症状无缓解自行停药。2024年8月出现双侧耳廓肿痛(图 2),伴鼻梁部疼痛,服塞来昔布胶囊0.2 g每日2次治疗,耳廓肿痛较前缓解,皮下结节仍反复,为明确诊断和治疗收入北京大学人民医院。近4个月体重下降2 kg。患者30年前有阑尾炎手术史。入院体检示:生命体征正常,贫血貌,心、肺、腹部查体未见明显异常,右踝关节内侧局部皮肤硬肿,触痛阳性。
辅助检查:白细胞1.1×109/L,中性粒细胞0.4×109 /L,淋巴细胞0.7×109 /L,血红蛋白66 g/L,平均红细胞压积100.2 fL,血小板58×109 /L,网织红细胞2.61%;血清铁及叶酸未见异常,总铁结合力稍低(49.95 μmol/L),铁蛋白790 μg/L,维生素B12 >2 000.0 ng/L;肝肾功能、电解质、凝血功能均未见异常;甲状腺功能未见异常;ESR 123 mm/h;CRP 138.9 mg/L;白介素-6(interleukin-6,IL-6)31.98 ng/L。降钙素原、乙肝五项、丙肝抗体、艾滋病病毒抗原抗体、梅毒血清特异性抗体、EB病毒DNA、巨细胞病毒DNA、结核感染T细胞检测均阴性。免疫球蛋白G 19.5 g/L,补体C4 0.54 g/L,血/尿免疫固定电泳阴性;抗人球蛋白试验阴性;ANA、抗ENA抗体谱、抗双链DNA、ANCA均阴性;冷球蛋白测定阴性;抗心磷脂抗体25.56 RU/mL;抗β2-糖蛋白Ⅰ抗体40.87 RU/mL;狼疮抗凝物测定阴性;抗磷脂酰丝氨酸-凝血酶原IgM抗体>400 RU/mL;抗β2-糖蛋白Ⅰ(结构域Ⅰ)抗体阴性。心脏彩色多普勒超声检查未见异常。肺、腹、盆部CT增强扫描未见明显异常。下肢血管彩色多普勒超声检查示双侧下肢动脉轻度硬化,双侧下肢静脉未见明显栓塞表现。骨髓涂片示骨髓增生Ⅲ级,可见粒细胞系统空泡变性,粒细胞与红细胞比值为3.81 ∶ 1;血小板散在可见。骨髓流式细胞学检测未见明显异常。骨髓增生异常综合征荧光原位杂交及DNA测序阴性。高分辨染色体核型分析示46, XY。骨髓病理组化检测示造血组织增生活跃,巨核细胞系统可见病态造血。外周血UBA1基因Exon3突变检测阳性,突变位点为p.Met41Val(c.121A>G)。
根据患者中年男性,慢性病程,表现为反复皮下痛性结节起病,后出现耳鼻软骨炎,查全血细胞减少,大细胞性贫血,ESR、CRP炎性指标明显升高,皮肤病理活检提示中性粒细胞性皮肤病改变,骨髓穿刺提示粒细胞系统空泡变性,外周血UBA1基因突变,故诊断为VEXAS综合征,主要累及耳鼻软骨、血液系统及皮肤,结合血液科会诊诊断骨髓增生异常综合征证据不足;同时,患者多个抗磷脂抗体阳性,筛查未见血栓证据,抗磷脂综合征诊断证据不足。治疗方面,2024年8月19日给予泼尼松50 mg每日1次口服,丙种球蛋白20 g每日1次静脉滴注3 d,利可君及达那唑胶囊对症升血细胞,辅以预防性抗感染、保护胃黏膜、补钙等综合治疗。2024年9月2日复查ESR 38 mm/h,CRP 0.6 mg/L,白细胞3.1×109 /L,红细胞2.5×1012 /L,血红蛋白90 g/L,平均红细胞压积100.2 fL,血小板79×109 /L,患者耳廓肿痛、四肢痛性结节缓解,炎症指标明显降低,因血小板计数低未加用抗血小板治疗。泼尼松逐渐规律减量,复查红细胞、血红蛋白及血小板明显恢复,白细胞仍低,2025年4月17日泼尼松减至20 mg/d时,复查白细胞2.18×109 /L,血红蛋白110 g/L,平均红细胞压积101.2 fL,血小板180×109 /L;ESR 6 mm/h,CRP 0.6 mg/L;抗心磷脂抗体、抗β2-糖蛋白Ⅰ抗体转为阴性,抗磷脂酰丝氨酸-凝血酶原IgM抗体35.6 RU/mL,炎性指标改善,磷脂抗体滴度较前明显下降,因白细胞仍低,为加强治疗及辅助激素减量加用磷酸芦可替尼片5 mg每日2次,充分告知血栓风险,联合小剂量阿司匹林预防性抗血小板治疗。复查血常规明显改善,泼尼松逐渐减至10 mg,每日1次,经治疗患者病情逐渐得到控制,随访至2025年4月,症状无反复。
2 讨论
VEXAS综合征于2020年首次被Beck等[1]报道,应用Sanger测序发现造血祖细胞中泛素样修饰激活酶1(ubiquitin-like modifier activating enzyme 1,UBA1)的体细胞突变,应用本病关键特征空泡(vacuoles)、E1酶(E1 enzyme)、X连锁(X-linked)、自身免疫性炎症(autoinflammatory)、体细胞(somatic) 的英文首字母命名。UBA1基因编码的E1酶是泛素-蛋白酶体系统的关键酶,在调节细胞稳态和蛋白稳态中发挥重要作用。体细胞和(或)骨髓祖细胞突变后导致细胞缺乏泛素化,这会增加细胞应激,刺激异常细胞因子的产生,并激活先天免疫途径,进而引发系列炎症级联反应和导致血液学异常[2]。因UBA1是X连锁基因,位于X染色体短臂(Xp11.3),使其可逃脱X染色体失活[2],VEXAS综合征多累及50岁以上男性,女性可受到未突变等位基因的保护,发病率远低于男性。通过Sanger测序分析可检测导致UBA1的突变,突变主要发生于p.Met-41位点,常见p.Met41Thr(c.122T>C)、p.Met41Val(c.121A>G)和p.Met41Leu(c.121A>C)共3种突变类型。本例患者UBA1基因突变位点是p.Met41Val(c.121A>G)。
目前VEXAS综合征的诊断尚依赖于典型的临床表现、实验室检查和遗传学检测。2025年美国风湿病学会(American College of Rheumatology,ACR)提出可通过外周血细胞和(或)骨髓样本中UBA1基因突变,骨髓活检显示髓系、红细胞系统前体细胞中存在空泡或大细胞性贫血、血小板减少等血液系统受累表现,以及器官或系统的反复炎症反应来诊断[3]。几乎所有病例都存在非特异性血液学异常,如贫血、血小板减少症或白细胞减少症、骨髓早期红细胞系统或髓系前体细胞中出现空泡,其他血液学特殊表现可能是由于UBA1基因突变,导致泛素-蛋白酶体系统功能紊乱,使机体对淋巴增殖性疾病及血液系统恶性肿瘤的易感性增加。80%以上的VEXAS综合征患者有皮肤受累,超过60%的患者首发症状即出现皮肤病变[4],皮肤病变总体以Sweet综合征、皮肤血管炎皮损为主。VEXAS综合征患者常出现软骨炎表现,对比特发性复发性多软骨炎(idiopathic-relapsing polychondritis,I-RP)和VEXAS综合征并发复发性多软骨炎(VEXAS-relapsing polychondritis,VEXAS-RP)的临床特征,均可出现耳软骨炎、肋软骨炎、非侵蚀性关节炎、外周血炎症指标升高、深静脉血栓等;而VEXAS-RP则特殊表现出50岁以上男性患病率高,更易出现发热、皮肤受累、血液系统受累、肺部受累(非气道受累)、心脏受累、眼部病变、关节炎及明显升高的炎症指标,并且更易复发, 病死率明显升高。因此,临床上出现患RP的中年男性患者皮肤、血液系统、肺部浸润性病变、心脏受累及炎症指标明显升高时应警惕VEXAS-RP,应积极完善外周血和(或)骨髓样本UBA1基因检测、骨髓穿刺等,更早识别VEXAS综合征。
此外,在多项队列研究中发现VEXAS综合征患者出现中性粒细胞驱动的涉及各级大小血管的一系列血管炎表型,还有合并ANCA阳性血管炎、合并全身广泛性血管炎出现复发性血栓性静脉炎至腹膜后出血死亡病例报道,提示血管炎可能是继发于VEXAS综合征的全身炎症反应表现[5-6]。相关研究报道VEXAS综合征患者血栓事件发生率高达40%~63%,血栓形成可能与心脏和肺部受累、活动性炎症状态、凝血因子活性增强、磷脂抗体阳性相关。合并血栓患者以静脉血栓栓塞为主,积极给予抗凝治疗后仍有部分患者反复发生血栓事件[7]。本例患者多项磷脂抗体阳性,应用激素联合磷酸芦可替尼片治疗目前暂未发生血栓事件,考虑积极控制患者全身炎症状态可能减少血管炎症,从而减少血栓栓塞发生,但需警惕患者血栓事件发生。
针对VEXAS综合征全身性、复发性的特点,治疗方案也应个体化、多样化,目前尚缺乏特定有效的治疗药物和治疗方式,主要为阻断炎症级联反应以及靶向和根除UBA1突变细胞。糖皮质激素是治疗VEXAS综合征首选药物,多数患者为避免病情复发,长期依赖大剂量激素治疗,但激素副作用亦增加了患者的负担。为辅助激素减量,根据其他自身炎症性疾病的治疗经验,临床报道应用Janus激酶(janus kinase,JAK)抑制剂、IL-6受体拮抗剂(托珠单抗)、白介素-1受体拮抗剂(阿那白滞素、卡那单抗)能在短时间内控制患者的全身炎症反应、减少病情复发,但减停激素并不常见[1, 8-11]。临床选择应根据病情择优使用,其中JAK抑制剂在症状缓解、控制炎症、激素减量、皮肤受累、血常规改善方面的疗效方面芦可替尼优于托法替尼、巴瑞替尼、乌帕替尼[9],但需注意其血栓栓塞、感染、肠穿孔风险。传统免疫抑制剂尚未表现出显著的辅助激素减量作用,对VEXAS综合征反应欠佳。对于血液系统受累,阿扎胞苷在治疗VEXAS综合征合并骨髓增生异常综合征案例报道中,临床和血液学疗效为46%~75%,并实现了激素减量[12-13]。根除骨髓中突变UBA1基因的唯一治疗是异基因造血干细胞移植(allogeneic hematopoietic stem cell transplantation,allo-HSCT),在回顾性分析时确诊为VEXAS综合征的allo-HSCT患者,在随访患者40个月时依然处于疾病缓解状态[14],但不适用于合并严重基础疾病和高龄患者。本例患者选择激素联合磷酸芦可替尼治疗,血常规及炎症改善,未发生血栓事件,并实现激素逐渐减量,随访至2025年4月未见复发。
总之,对于50岁以上的男性软骨炎患者,出现顽固性自身炎症表现和(或)难以解释的血液系统异常时,应重视骨髓评估,进行UBA1基因检测,警惕VEXAS综合征可能,尽早明确诊断、制定个性化治疗方案有助于改善患者预后,并需密切随访。

{kind=link}
{kind=link}
{kind=link}
{kind=link}