
特发性低促性腺激素性腺功能减退症(idiopathic hypogonadotropic hypogonadism,IHH)是一种罕见病,由于下丘脑、垂体分泌激素异常,导致促性腺激素水平低下,性腺功能减退,性激素分泌不足,从而出现青春期发育延迟和第二性征发育异常。女性临床表现主要为原发性闭经、乳腺不发育、幼稚外阴等,男性则主要表现为隐睾、小阴茎、不育,此外,还可出现嗅觉异常、唇腭裂、骨骺闭合延迟、镜像运动和听力障碍等其他临床表现。根据患者是否存在嗅觉异常,IHH可进一步分类为卡尔曼综合征(Kallmann syndrome,KS)及非嗅觉异常IHH(normosmic IHH,nIHH)。值得关注的是,该疾病多见于男性,男性患病率为1/30 000至1/10 000,约为女性的3~5倍[1],但部分研究认为,由于女性青春期才开始出现相应症状,临床表现为第二性征发育不全、闭经,生殖系统表型轻重程度差异大,其他系统无特异性表型,导致临床上难以早期诊断且容易漏诊,因此,女性IHH的患病率可能被低估,其实际患病率及其遗传学背景仍需进一步研究[2]。
目前发现的该疾病风险基因已超过50个,人类孟德尔遗传数据库(Online Mendelian Inheritance in Man,OMIM)收录了27个,其中在人群中检出率较高的基因包括FGFR1 (9.8%)、CHD7 (5.4%)、PROKR2 (6.1%)等,但患者整体变异检出率仅为30%[3],当前对该疾病的致病机制及风险基因仍未完全阐明。同时,该疾病的遗传模式除较为常见的单基因遗传,还存在双基因遗传、寡基因遗传模式,即同时携带两个及以上基因异常才出现相应表型。其中,我们关注信号素3A(semaphorin 3A,SEMA3A)调节嗅觉受体神经元轴突引导过程及促性腺激素释放激素(gonadotropin-releasing hormone,GnRH)神经元的迁移过程,该迁移过程与丛状蛋白相关,文献证明SEMA3A缺失会导致GnRH神经元迁移过程出现异常,从而导致促性腺激素分泌不足,出现性腺功能减退。丛状蛋白A1(PLXNA1)作为SEMA3A等蛋白的主要受体蛋白复合体成员,对于轴突导向、细胞侵袭性生长及细胞迁移具有重要作用,已有文献证明PLXNA1缺失也会导致GnRH神经元迁移异常,性腺功能发育异常[4],且该蛋白与PLXNA3的联合缺失可导致低促性腺激素的表现[5]。PLXNA1在IHH患者中的变异携带率约为4%,其基因型-表型关联尚未完全阐明。本研究聚焦女性IHH患者的PLXNA1变异携带情况,以进一步了解其临床表现及遗传学特点。
1 资料与方法
1.1 研究对象
招募2022年1月至2023年6月就诊于北京大学第三医院的IHH女性患者。纳入标准:(1)16岁月经未来潮或原发性闭经;(2)第二性征不发育或发育不完全;(3)促性腺激素(卵泡刺激素、黄体生成素)水平低下或正常,雌二醇≤20 ng/L。排除标准:继发性闭经及功能性闭经,如垂体腺瘤、服用抑制性腺轴药物、患有系统性疾病(如多囊卵巢综合征等)。本研究已通过北京大学第三医院医学科学研究伦理委员会批准(批准号:M2022001),所有参与者均已签署知情同意书。
1.2 临床资料收集
收集患者病史资料,包括:(1)青春期发育年龄、第二性征发育情况(Tanner分期)、月经情况、治疗前激素水平、子宫及附件发育情况、用药史及辅助生殖治疗情况;(2)嗅觉检查、头颅磁共振成像检查及其他辅助检查;(3)婚育史及家族史等。
根据患者是否存在嗅觉异常将患者分为KS组及nIHH组,比较两组年龄、体重指数(body mass index, BMI)及基础性激素水平。
1.3 全外显子组检测及Sanger验证
采集患者及其家系成员4 mL外周血样本,置于含有乙二胺四乙酸(ethylenediaminetetraacetic acid,EDTA)抗凝剂的抽血管中,通过试剂盒法提取外周血基因组DNA,构建基因文库进行全外显子组测序,对检出的变异位点进行Sanger测序验证及家系验证。
1.4 统计学方法
使用SPSS 26.0软件进行统计学分析。对连续变量进行正态性检验(Kolmogorov-Smirnov),若符合正态分布则用${\bar x}$±s表示,采用Student’ s t检验进行组间比较;若不符合正态分布则用M(P25,P75)表示,采用Mann-Whitney检验进行组间比较。双侧检验,P < 0.05为差异有统计学意义。
2 结果
2.1 患者临床基本资料
本研究共招募了21例女性患者,均因出现原发性闭经、青春期第二性征未发育而就诊,包括15例nIHH和6例KS,其中4个家系存在相关家族史。对比KS组与nIHH组基线资料,其年龄、体重指数、促性腺激素和性激素水平差异均无统计学意义(表 1)。21例患者均表现为低促性腺激素和低雌激素水平,尤其是卵泡刺激素、黄体生成素显著低下。除6例KS患者存在嗅觉异常外,未发现任何患者存在其他系统异常(如唇腭裂、肾脏发育不良、心脏发育畸形、骨骼/牙齿发育异常、听力异常等)。
表1 21例女性IHH患者临床资料Table 1 Clinical data of 21 female IHH patients |
| Items | nIHH (n=15) | KS (n=6) | Statistics | P |
| Age/years | 31.0 (25.0, 33.0) | 27.0 (22.5, 33.3) | Z=-0.981 | 0.326 |
| BMI/(kg/m2) | 21.43 (19.93, 22.48) | 23.84 (21.77, 25.38) | Z=-1.815 | 0.070 |
| FSH/(IU/L) | 0.55 (0.25, 1.87) | 0.75 (0.22, 2.57) | Z=-0.078 | 0.938 |
| LH/(IU/L) | 0.23 (0.11, 0.73) | 0.41 (0.10, 0.71) | Z=-0.078 | 0.938 |
| E2/(ng/L) | 12.00 (10.00, 26.40) | 14.80 (9.98, 21.98) | Z=-0.397 | 0.692 |
| P/(μg/L) | 0.70 (0.11, 0.87) | 0.83 (0.35, 1.83) | Z=-1.092 | 0.275 |
| PRL/(μg/L) | 10.05±18.86 | 18.88±25.92 | t=-0.872 | 0.394 |
| T/(ng/dL) | < 0.69 | < 0.69 |
Data are expressed as ${\bar x}$±s or M (P25, P75).IHH, idiopathic hypogonadotropic hypogonadism; nIHH, normosmic IHH; KS, Kallmann syndrome; BMI, body mass index; FSH, follicle-stimulating hormone; LH, luteinizing hormone; E2, estradiol; P, progesterone; PRL, prolactin; T, testosterone. |
2.2 遗传学检测结果
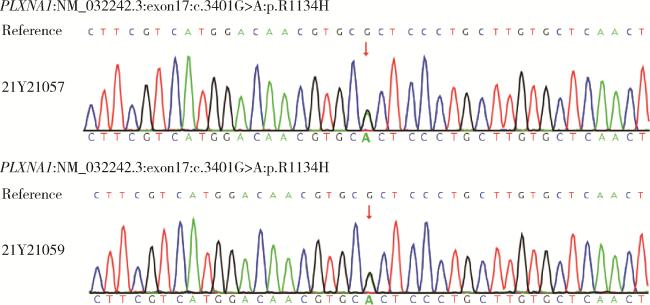
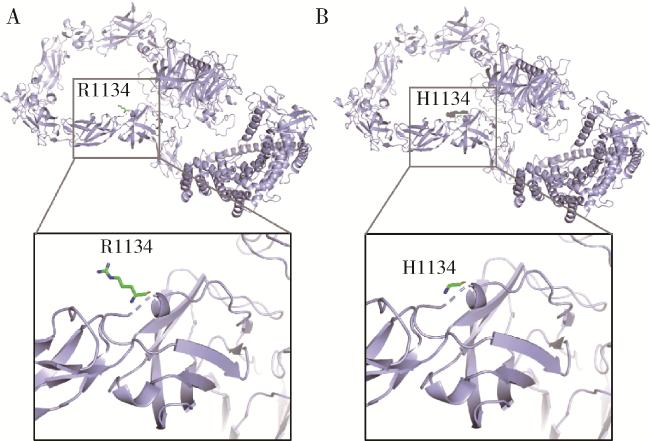
17例患者进行了全外显子组基因检测,其中4例(23.5%)检出明确致病性基因变异,涉及FGFR1及PROKR2基因。对其余13例患者进行重分析及变异位点筛选,发现2例KS患者携带PLXNA1基因相同变异位点PLXNA1 : c.3401G>A(p.R1134H),该变异位点的美国医学遗传学与基因组学学会(American College of Medical Genetics and Genomics,ACMG)评级为临床意义不明(PM2+PP2),Sanger验证表明2例均杂合携带该变异位点(图 1)。由于未获得这2例患者父母的血液样本,故无法进行变异来源分析。利用Alphafold2对PLXNA1错义变异进行蛋白质结构预测(图 2),该变异导致第1134号氨基酸由精氨酸变为组氨酸,氨基酸疏水性发生改变,提示该变异对SEMA3A-PLXNA1配体-受体结合可能存在影响。
2.3 女性IHH患者遗传特点及辅助生殖临床治疗
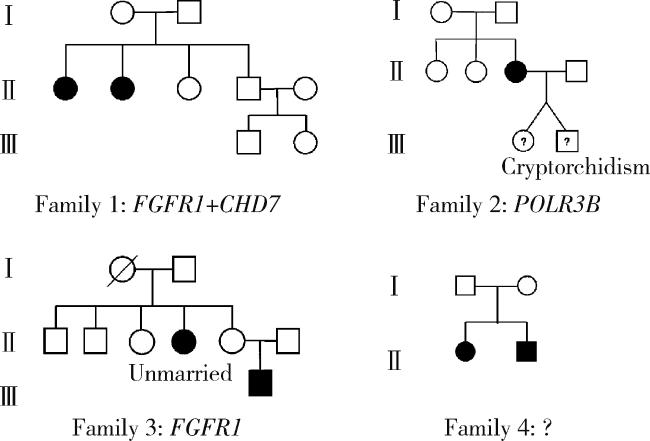
21例患者中4例存在相关家族史,家系图见图 3。由于表型-基因型不完全相符、不符合共分离、无相关致病性变异位点等原因,4个家系均未获得明确遗传学诊断。家系1有两名女性(nIHH)携带FGFR1及CHD7变异位点,均为临床意义不明,存在第二性征发育不全及原发性闭经,嗅觉正常,无其他系统发育异常。家系2女性患者杂合携带POLR3B变异位点,该变异为常染色体隐性遗传;患者通过辅助生殖技术获得双胎妊娠,其中男胎儿出生时发现隐睾,已进行手术治疗。家系3先证者为男性,杂合携带FGFR1变异,其早期即已明确诊断,通过激素补充治疗维持第二性征发育及生精功能;先证者姑姑未治疗,至今未婚未育。家系4患者兄妹二人均存在第二性征发育不全,患者表现为原发性闭经,但未检出相关变异位点。以上结果提示即使IHH患者存在家族史,由于遗传模式复杂及涉及多个基因,其获得准确遗传诊断仍十分困难。
另外17例患者均为散发,其中9例未婚,当前仅通过药物治疗维持人工月经周期;4例已通过辅助生殖治疗生育后代,由于遗传学诊断尚未明确,当前不能判断子代是否存在患病风险;2例正处于辅助生殖助孕治疗过程中。
3 讨论
IHH表型异质性较高,其遗传学病因复杂,致病性变异基因检出率约为50%,且检出变异位点评级多为临床意义不明,存在多个基因变异共同导致疾病发生的可能。当前国内针对该疾病的研究集中在对FGFR1、KAL1、CHD7等较为常见的基因进行遗传学分析。FGFR1基因为常染色体显性遗传,携带率可达到12%[6],为检出率最高的基因,本研究中有3例患者携带FGFR1基因的致病性变异。KAL1基因为X连锁显性遗传,整体携带率为6%~7%,多见于男性,携带该变异的患者存在肾脏发育异常的可能[7-8]。CHD7基因同时与CHARGE综合征相关,可能同时出现多器官发育障碍,包括唇腭裂、听力异常、先天性心脏病、骨骼发育异常等[9]。
PLXNA1的致病性及遗传方式仍存在争议,目前国内外对于PLXNA1的研究有限,仅两项研究针对该基因进行了较为全面的遗传变异图谱的分析[10-11]。一项研究表明,Plxna1敲除会导致雄性小鼠GnRH神经元数量减少,生育能力降低,但该表型存在个体差异性,Plxna1敲除的雌鼠无显著表型,且生育力正常[4]。另一项研究表明,Plxna1单独敲除在E14.5小鼠并不会导致大脑嗅球区、筛板、前脑的GnRH神经元数目异常,但出生后小鼠的下丘脑内侧视前区GnRH神经元数量显著减少,Plxna1、Plxna3双基因缺失小鼠表型则更为严重[5]。目前认为PLXNA1变异导致的疾病表型较轻,当其他同家族受体或协同受体同时出现异常时可能导致表型加重,因此,对于该通路相关基因的变异筛查十分必要。关于PLXNA1基因,目前已报道了31个错义突变位点,可见于KS患者及nIHH患者,检出率约为3.9%~5.6%[10-11],变异位点包括:p.P40R、p.V287M、p.R319W、p.V349L、p.V437L、p.P485L、p.V491I、p.R528W、p.V536I、p.H684Y、p.G720E、p.R740H、p.R813H、p.R836H、p.R840Q、p.P848R、p.A854T、p.R897H、p.T1067M、p.A1106V、p.V1113L、p.R1134H、p.P1181L、p.A1210V、p.K1451R、p.L1464V、p.K1618T、p.I1701V、p.C1744F、p.S1709L和p.Ser1850Arg[4, 10-11],携带该基因变异的患者可表现为nIHH,也可表现为KS。
有研究曾报道1例携带PLXNA1 : c.3401G>A变异位点的散发KS男性患者,且该患者不携带其他相关致病性基因变异[10]。本研究报道的携带PLXNA1 : c.3401G>A错义变异的2例患者也全部表现为KS,无其他系统异常,且不携带SEMA3A变异位点,提示PLXNA1 : c.3401G>A变异位点可能为KS患者的风险变异位点。蛋白质结构预测结果提示,氨基酸改变影响单氨基酸疏水性,但氨基酸改变对配体与受体间的相互作用及对疾病表型的影响仍需进一步研究。PLXNA1 : c.3401G>A变异单独致病证据不足,不排除可能存在其他基因变异位点或未知致病基因导致KS的可能性。有研究进一步筛选了相关基因变异位点,其中SEMA3A已被证实为IHH致病基因之一(OMIM:614897),遗传模式为常染色体显性遗传,PLXNA1作为其主要受体,NRP1和NRP2作为其协同受体,共同参与调节GnRH神经元的迁移[12],但本研究中的2例KS患者均未检测到SEMA3A、PLXNA3、NRP1、NRP2相关变异。
IHH会导致青春期不发育或发育延迟,需长期用药以保证生殖器官及第二性征发育以获得生育能力,对患者的经济负担及心理压力都造成巨大的负担。由于当前对该疾病的了解不足,加之该疾病致病因素复杂、遗传模式多样,难以对患者进行明确的遗传学诊断,更无法对后代患病风险进行较为准确的评估。可以明确的是,该疾病存在多基因致病的情况,因此,若要分析其遗传学病因,需要更大的样本量以构建多基因风险评估模型并分析疾病遗传风险。另外,本研究中女性患者遗传变异检出率低于50%,且检出变异的部分家系的基因型-表型共分离情况尚未得到验证。同时,部分女性患者由于表型较轻或检查不充分而未能得到明确的遗传学诊断,但实际可能存在遗传风险,其遗传学背景及表型关联尚需进一步研究。
综上,PLXNA1 : c.3401G>A作为潜在KS风险变异位点,临床遗传检测时应关注家系成员是否携带同一变异及是否发病,评估遗传学病因及遗传风险。将来关于IHH的研究应通过扩大样本量,进一步收集患者详细家系信息及临床病史,以全面分析遗传背景,明确该疾病的遗传学病因,为遗传咨询提供更可靠的理论依据及遗传诊断方法。

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}