北京大学学报(医学版) ›› 2022, Vol. 54 ›› Issue (4): 751-755. doi: 10.19723/j.issn.1671-167X.2022.04.027
46XX-17α-羟化酶缺乏症助孕成功1例
张春梅1,杨蕊1,李蓉1,乔杰1,王海宁2,王颖1,*( )
)
- 1. 北京大学第三医院妇产科,生殖医学中心,北京 100191
2. 北京大学第三医院内分泌科,北京 100191
Successful assisted reproductive technology treatment for a woman with 46XX-17α-hydroxylase deficiency: A case report
Chun-mei ZHANG1,Rui YANG1,Rong LI1,Jie QIAO1,Hai-ning WANG2,Ying WANG1,*()
- 1. Center for Reproductive Medicine, Department of Obstetrics and Gynecology, Peking University Third Hospital, Beijing 100191, China
2. Department of Endocrinology, Peking University Third Hospital, Beijing 100191, China
摘要:
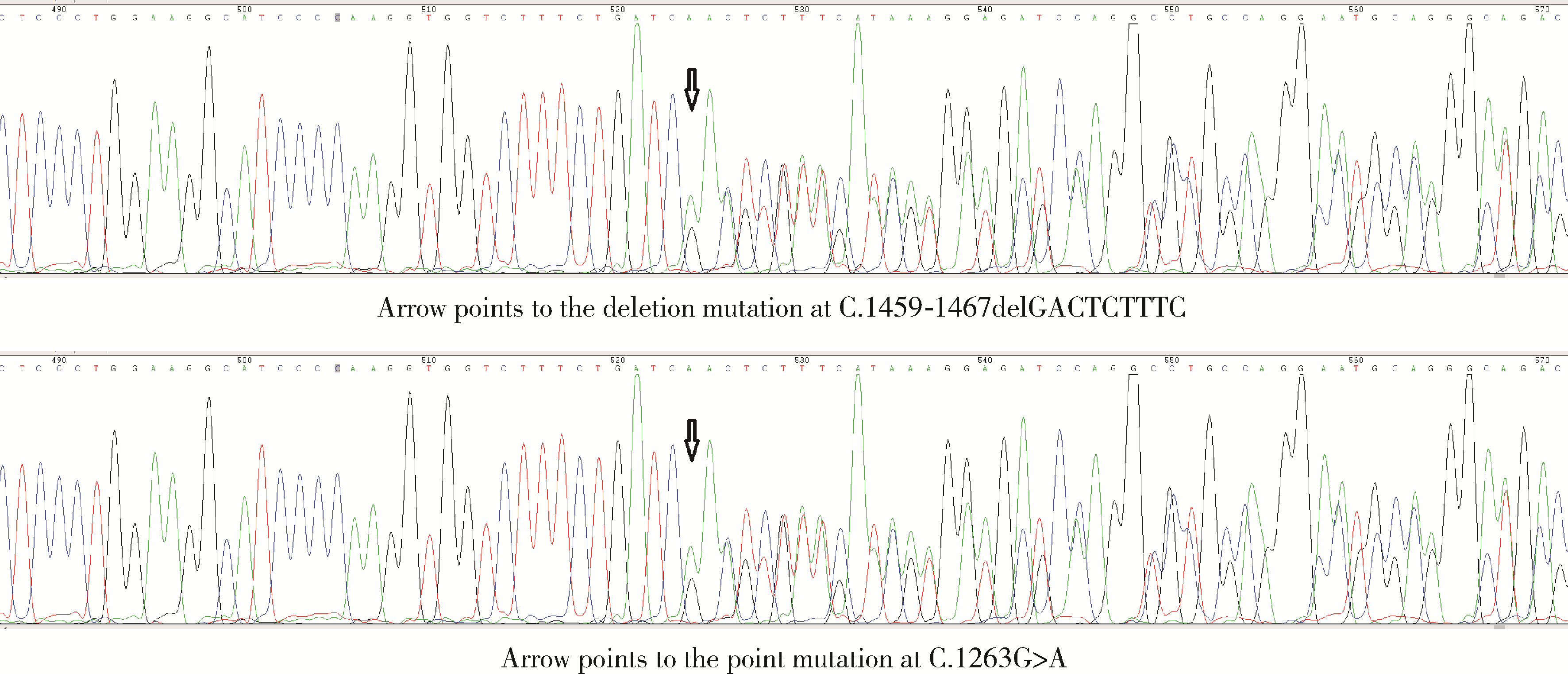
先天性肾上腺皮质增生症(congenital adrenal hyperplasia,CAH)是一类常染色体隐性遗传病,其中17α-羟化酶缺乏症(17α-hydroxylase deficiency,17α-OHD)为CAH少见类型。17α-OHD为CYP17基因突变所致,导致17α-羟化酶部分或完全缺乏,体内不能生成足够的皮质醇及性激素,表现为促肾上腺皮质激素(adrenocorticotropic hormone,ACTH)分泌过多,雌二醇(estradiol,E2)和雄激素降低,孕酮(progesterone,P)、促卵泡生成素(follicle stimulating hormone,FSH)和黄体生成素(luteinizing hormone,LH)升高。患者社会性别多为女性,按照染色体核型可分为46XX和46XY,其中46XX型更少见。临床表现为低血钾和高血压,46XX型患者可出现月经不规律、闭经和不孕。患者症状轻重可因17α-羟化酶缺乏程度不同而异,部分性17α-OHD患者因症状不典型,易漏诊或误诊。染色体核型为46XX的部分性17α-OHD患者可有不同程度内外生殖器官的发育和月经来潮,具备潜在排卵和生育机会,但由于体内高P水平对子宫内膜的不良影响,患者存在不孕的问题。目前关于46XX-17α-OHD助孕治疗的报道国外仅见4例,国内曾有文献报道2例,但未提及其助孕过程。北京大学第三医院生殖医学中心1例46XX-17α-羟化酶缺乏症患者经过体外受精-胚胎移植技术(in vitro fertilization-embryo transfer,IVF-ET)治疗后成功受孕并分娩。首先给予患者糖皮质激素治疗降低P水平,随后采用超长方案促排卵,获卵10枚,冻存胚胎6枚。促性腺激素释放激素激动剂(gonadotropin releasing hormone agonist,GnRH-a)降调节人工周期解冻移植后,患者成功妊娠并于妊娠37周剖宫产分娩1名健康男婴。本例患者的诊治过程提示,46XX-17α-OHD患者可以有良好的卵母细胞和胚胎,IVF联合人工周期解冻移植可以是此类患者助孕的有效方法。
中图分类号:
- R711.6

| 1 |
Marsh CA , Auchus RJ . Fertility in patients with genetic deficiencies of cytochrome P450c17 (CYP17A1): Combined 17-hydroxylase/17, 20-lyase deficiency and isolated 17, 20-lyase deficiency[J]. Fertil Steril, 2014, 101 (2): 317- 322.
doi: 10.1016/j.fertnstert.2013.11.011 |
| 2 |
Ben-Nun I , Siegal A , Shulman A , et al. Induction of artificial endometrial cycles with oestradiol implants and injectable progesterone: Establishment of a viable pregnancy in a woman with 17-alpha-hydroxylase deficiency[J]. Hum Reprod, 1995, 10 (9): 2456- 2458.
doi: 10.1093/oxfordjournals.humrep.a136319 |
| 3 | 程炜, 孙爱军. 17α羟化酶缺乏症(46, XX)病例讨论[J]. 实用妇科内分泌电子杂志, 2016, 3 (5): 16- 19. |
| 4 |
郁琦, 王含必, 何方方. 17α羟化酶缺乏症13例临床分析[J]. 中国实用妇科与产科杂志, 2004, 20 (11): 656- 658.
doi: 10.3969/j.issn.1005-2216.2004.11.010 |
| 5 |
金楠, 巴建明. 先天性肾上腺皮质增生症的药物治疗[J]. 药品评价, 2013, 10 (7): 15- 20.
doi: 10.3969/j.issn.1672-2809.2013.07.006 |
| 6 | 郝凤梅, 田秦杰, 郁琦. 染色体为46, XX的17α羟化酶缺乏症8例临床分析[J]. 中国实用妇科与产科杂志, 2008, 24 (12): 934- 935. |
| 7 | 黄艳, 朱颖, 王可, 等. 促性腺激素垂体腺瘤引起卵巢过度刺激一例并文献复习[J]. 中国妇产科临床杂志, 2018, 19 (4): 362- 363. |
| 8 |
刘亚萌, 夏艳洁, 李小英, 等. 17α-羟化酶/17, 20-裂解酶缺陷症六例分析[J]. 中华内分泌代谢杂志, 2019, 35 (10): 825- 828.
doi: 10.3760/cma.j.issn.1000-6699.2019.10.002 |
| 9 |
Bianchi PH , Gouveia GR , Costa EM , et al. Successful live birth in a woman with 17alpha-hydroxylase deficiency through IVF frozen-thawed embryo transfer[J]. J Clin Endocrinol Metab, 2016, 101 (2): 345- 348.
doi: 10.1210/jc.2015-3201 |
| 10 | Falhammar H . Successful fertility outcome in a woman with 17α-hydroxylase deficiency[J]. Clin Endocrinol (Oxf), 2018, 88 (4): 607- 609. |
| 11 |
Kitajima M , Miura K , Inoue T , et al. Two consecutive successful live birth in woman with 17alpha hydroxylase deficiency by frozen-thaw embryo transfer under hormone replacement endometrium preparation[J]. Gynecol Endocrinol, 2018, 34 (5): 381- 384.
doi: 10.1080/09513590.2017.1393512 |
| 12 |
Carvalho LC , Brito VN , Martin RM , et al. Clinical, hormonal, ovarian, and genetic aspects of 46, XX patients with congenital adrenal hyperplasia due to CYP17A1 defects[J]. Fertil Steril, 2016, 105 (6): 1612- 1619.
doi: 10.1016/j.fertnstert.2016.02.008 |
| 13 | 张琳, 王海宁, 洪天配. 17α-羟化酶缺乏症一例的诊治经验及文献回顾[J]. 北京大学学报(医学版), 2008, 40 (2): 221- 222. |
| [1] | 沈浣, 田莉, 刘斌. 不孕及反复自然流产患者小卵泡排卵的诊治意义[J]. 北京大学学报(医学版), 2003, 35(2): 166-169. |
|
||
