Journal of Peking University (Health Sciences) ›› 2021, Vol. 53 ›› Issue (5): 957-963. doi: 10.19723/j.issn.1671-167X.2021.05.025
Previous Articles Next Articles
Clinical, pathological and genetic characteristics of 8 patients with distal hereditary motor neuropathy
LIU Mei-ge1,FANG Pu2,WANG Yan1,CONG Lu1,FAN Yang-yi1,YUAN Yuan1,XU Yan1,ZHANG Jun1,HONG Dao-jun1,2,△( )
)
- 1. Department of Neurology, Peking University People’s Hospital, Beijing 100044, China
2. Department of Neurology, the First Affiliated Hospital of Nanchang University, Nanchang 330006, China
CLC Number:
- R596


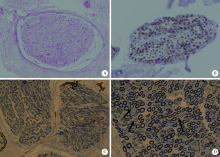
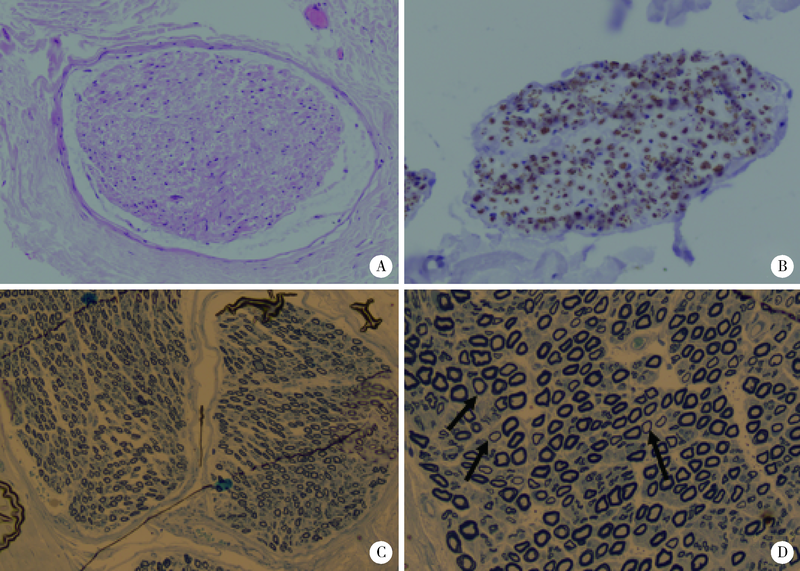
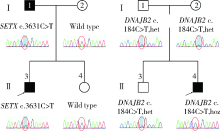

| [1] |
Garg N, Park SB, Vucic S, et al. Differentiating lower motor neuron syndrome [J]. J Neurol Neurosurg Psychiatry, 2017, 88(6):474-483.
doi: 10.1136/jnnp-2016-313526 |
| [2] |
Frasquet M, Rojas-García R, Argente-Escrig H, et al. Distal hereditary motor neuropathies: mutation spectrum and genotype-phenotype correlation [J]. Eur J Neurol, 2021, 28(4):1334-1343.
doi: 10.1111/ene.14700 pmid: 33369814 |
| [3] |
Bansagi B, Griffin H, Whittaker RG, et al. Genetic heterogeneity of motor neuropathies [J]. Neurology, 2017, 88(13):1226-1234.
doi: 10.1212/WNL.0000000000003772 |
| [4] | Bacquet J, Stojkovic T, Boyer A, et al. Molecular diagnosis of inherited peripheral neuropathies by targeted next-generation sequencing: molecular spectrum delineation [J]. BMJ Open, 2018, 8(10):e21632. |
| [5] |
Echaniz-Laguna A, Geuens T, Petiot P, et al. Axonal neuropathies due to mutations in small heat shock proteins: clinical, genetic, and functional insights into novel mutations [J]. Hum Mutat, 2017, 38(5):556-568.
doi: 10.1002/humu.23189 pmid: 28144995 |
| [6] |
Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology [J]. Genet Med, 2015, 17(5):405-424.
doi: 10.1038/gim.2015.30 pmid: 25741868 |
| [7] |
Tanabe H, Higuchi Y, Yuan JH, et al. Clinical and genetic features of charcot-marie-tooth disease 2F and hereditary motor neuropathy 2B in Japan [J]. J Peripher Nerv Syst, 2018, 23(1):40-48.
doi: 10.1111/jns.2018.23.issue-1 |
| [8] |
Windpassinger C, Auer-Grumbach M, Irobi J, et al. Heterozygous missense mutations in BSCL2 are associated with distal hereditary motor neuropathy and Silver syndrome [J]. Nat Genet, 2004, 36(3):271-276.
doi: 10.1038/ng1313 |
| [9] |
Novarino G, Fenstermaker AG, Zaki MS, et al. Exome sequencing links corticospinal motor neuron disease to common neurodegenerative disorders [J]. Science, 2014, 343(6170):506-511.
doi: 10.1126/science.1247363 pmid: 24482476 |
| [10] | Xie Y, Lin Z, Pakhrin PS, et al. Genetic and clinical features in 24 Chinese distal hereditary motor neuropathy families [J/OL]. Front Neurol, 2020, 11:603003(2020-12-14)[2020-12-15]. https://pubmed-ncbi-nlm-nih-gov-443.webvpn.bjmu.edu.cn/33 3810781/ . |
| [11] | 张付峰, 卢晓琴, 严新翔, 等. 远端型遗传性运动神经病的临床特征分析 [J]. 第二军医大学学报, 2009, 30(1):57-60. |
| [12] |
De Jonghe P, Auer-Grumbach M, Irobi J, et al. Autosomal dominant juvenile amyotrophic lateral sclerosis and distal hereditary motor neuronopathy with pyramidal tract signs: synonyms for the same disorder [J]. Brain, 2002, 125(Pt 6):1320-1325.
pmid: 12023320 |
| [13] |
Motley WW, Griffin LB, Mademan I, et al. A novel AARS mutation in a family with dominant myeloneuropathy [J]. Neurology, 2015, 84(20):2040-2047.
doi: 10.1212/WNL.0000000000001583 |
| [14] |
Luigetti M, Fabrizi GM, Madia F, et al. Seipin S90L mutation in an Italian family with CMT2/dHMN and pyramidal signs [J]. Muscle Nerve, 2010, 42(3):448-451.
doi: 10.1002/mus.21734 pmid: 20806400 |
| [15] |
Beecroft SJ, McLean CA, Delatycki MB, et al. Expanding the phenotypic spectrum associated with mutations of DYNC1H1 [J]. Neuromuscul Disord, 2017, 27(7):607-615.
doi: 10.1016/j.nmd.2017.04.011 |
| [16] |
Dierick I, Baets J, Irobi J, et al. Relative contribution of mutations in genes for autosomal dominant distal hereditary motor neuropathies: a genotype-phenotype correlation study [J]. Brain, 2008, 131(Pt 5):1217-1227.
doi: 10.1093/brain/awn029 pmid: 18325928 |
| [17] |
Rossor AM, Evans MR, Reilly MM. A practical approach to the genetic neuropathies [J]. Pract Neurol, 2015, 15(3):187-198.
doi: 10.1136/practneurol-2015-001095 |
| [18] |
Liu X, Duan X, Zhang Y, Sun A, et al. Molecular analysis and clinical diversity of distal hereditary motor neuropathy [J]. Eur J Neurol, 2020, 27:1319-1326.
doi: 10.1111/ene.14260 pmid: 32298515 |
| [19] |
Beijer D, Baets J. The expanding genetic landscape of hereditary motor neuropathies [J]. Brain, 2020, 143(Pt 12):3540-3563.
doi: 10.1093/brain/awaa311 |
| [1] | Jingxian WU, Liuyan ZHENG, Huan YU, Huairong WANG, Shuting XIE, Yalin CHEN, Teng LI, Mengying WANG, Xueying QIN, Tao WU, Dafang CHEN, Yiqun WU, Yonghua HU. Association analysis between genetic nurturing effects of CTNNA gene family and ischemic stroke [J]. Journal of Peking University (Health Sciences), 2026, 58(3): 528-535. |
| [2] | Hongjuan BAI, Yang CHEN, Kun WANG, Xuebing XU. Relationship between family trauma, school bullying and suicidal behavior in adolescents: Regulatory effect of DRD2 gene polymorphism [J]. Journal of Peking University (Health Sciences), 2026, 58(3): 606-615. |
| [3] | Hai WANG, Yizhou JIANG. Molecular mechanisms and clinical applications of anti-angiogenic therapy in precision treatment of breast cancer [J]. Journal of Peking University (Health Sciences), 2026, 58(2): 251-256. |
| [4] | Zheng LI, Longwei LV, Xiao ZHANG, Dandan XIA, Ping ZHANG, Yunsong LIU, Yongsheng ZHOU. Advances in oral and craniofacial bone regeneration modulated by stem cells and biomaterials [J]. Journal of Peking University (Health Sciences), 2026, 58(2): 272-277. |
| [5] | Han ZHANG, Fujia YANG, Ruili YANG. Progress in regulating stem cell functions for repair and regeneration of craniomaxillofacial tissues [J]. Journal of Peking University (Health Sciences), 2026, 58(2): 285-289. |
| [6] | Xinying WANG, Xueyuan CHENG, Mengjun ZHANG, Fei LI, Jinyu DUAN, Jing QIAO. Effect of concentrated growth factors in guided tissue regeneration for the treatment of mandibular molar furcation lesions [J]. Journal of Peking University (Health Sciences), 2026, 58(2): 372-379. |
| [7] | Haoming YIN, Zijie WANG, Fan SHU, Zhanyi ZHANG, Hui LIANG, Shudong ZHANG. Expression and significance of the FABP6 long transcript in clear cell renal cell carcinoma [J]. Journal of Peking University (Health Sciences), 2026, 58(2): 393-398. |
| [8] | Haifeng DONG, Hengxing CHEN, Changhua ZHANG. Research progress on protein lactylation modification in malignant tumors [J]. Journal of Peking University (Health Sciences), 2026, 58(2): 423-430. |
| [9] | Lingfu ZHANG, Ming CHEN, Xiaoyu ZHAO, Gang WANG, Long CUI, Xiaofeng LING, Lixin WANG, Zhi XU, Limei GUO, Chunsheng HOU. Gross classification of gallbladder cancer with primary lesion limited to the gallbladder wall and its correlation with prognosis and precancerous lesions [J]. Journal of Peking University (Health Sciences), 2026, 58(1): 184-189. |
| [10] | Yunling GENG, Chao LIU, Ping YANG, Jiajia ZHENG, Ning SHEN, Yipeng DU. Clinical features and virulence gene distribution of Klebsiella pneumoniae multi-site infection in patients with hospital-acquired pneumonia [J]. Journal of Peking University (Health Sciences), 2026, 58(1): 201-207. |
| [11] | Peng BAI, Hao ZHANG, Jiechu WANG, He ZHU, Hong ZENG. Perioperative management of a patient with Kennedy disease undergoing knee replacement: A case report [J]. Journal of Peking University (Health Sciences), 2026, 58(1): 225-227. |
| [12] | Wenbo LI, Yufeng SHEN, Yongtao YANG, Shenyao SHAN, Zixiang GAO, Aonan WEN, Xiangyi SHANG, Yuwen TIAN, Shuwei GUO, Yizhen WANG, Yong WANG, Yijiao ZHAO. Development of a surface electromyography index system for orofacial muscles and validation of a discriminant model in unilateral molar occlusal interference [J]. Journal of Peking University (Health Sciences), 2026, 58(1): 89-98. |
| [13] | Yanhua LIU, Min LU, Xuyang ZHAO, Kuan'gen ZHANG, Rui WU, Fang MEI, Zhihao DAI, Jiangfeng YOU, Fei PEI. Effect of dephosphorylation of tumor metastasis suppressor gene LASS2 on vacuolar ATPase activity and invasiveness of prostate cancer [J]. Journal of Peking University (Health Sciences), 2025, 57(6): 1113-1123. |
| [14] | Qi DONG, Jing HE, Yuan JIA, Haihong YAO, Xia ZHANG. VEXAS syndrome mimicking relapsing polychondritis: A case report [J]. Journal of Peking University (Health Sciences), 2025, 57(6): 1180-1183. |
| [15] | Xiaolin WANG, Luyao LI, Wen ZHANG, Hongyan WANG. Clinicopathological analysis of mesonephric-like adenocarcinoma in the corpusuteri: A report of 3 cases [J]. Journal of Peking University (Health Sciences), 2025, 57(6): 1208-1212. |
|
||