北京大学学报(医学版) ›› 2026, Vol. 58 ›› Issue (2): 327-331. doi: 10.19723/j.issn.1671-167X.2026.02.016
17例女性特发性低促性腺激素性腺功能减退症患者遗传变异分析
陈琪琪1, 王海宁2, 刘烨2, 智旭1,*( )
)
- 1. 北京大学第三医院妇产科,生殖医学中心;女性生育力促进全国重点实验室;国家妇产疾病临床医学研究中心(北京大学第三医院);辅助生殖教育部重点实验室(北京大学);生殖内分泌与辅助生殖技术北京市重点实验室,北京 100191
2. 北京大学第三医院内分泌科,北京 100191
Genetic variants analysis of 17 female patients with idiopathic hypogonadotropic hypogonadism
Qiqi CHEN1, Haining WANG2, Ye LIU2, Xu ZHI1,*()
- 1. Department of Obstetrics and Gynecology, Center for Reproductive Medicine, Peking University Third Hospital; State Key Laboratory of Female Fertility Promotion; National Clinical Research Center for Obstetrics and Gynecology (Peking University Third Hospital); Key Laboratory of Assisted Reproduction (Peking University), Ministry of Education; Beijing Key Laboratory of Reproductive Endocrinology and Assisted Reproductive Technology, Beijing 100191, China
2. Department of Endocrinology and Metabolism, Peking University Third Hospital, Beijing 100191, China
摘要:
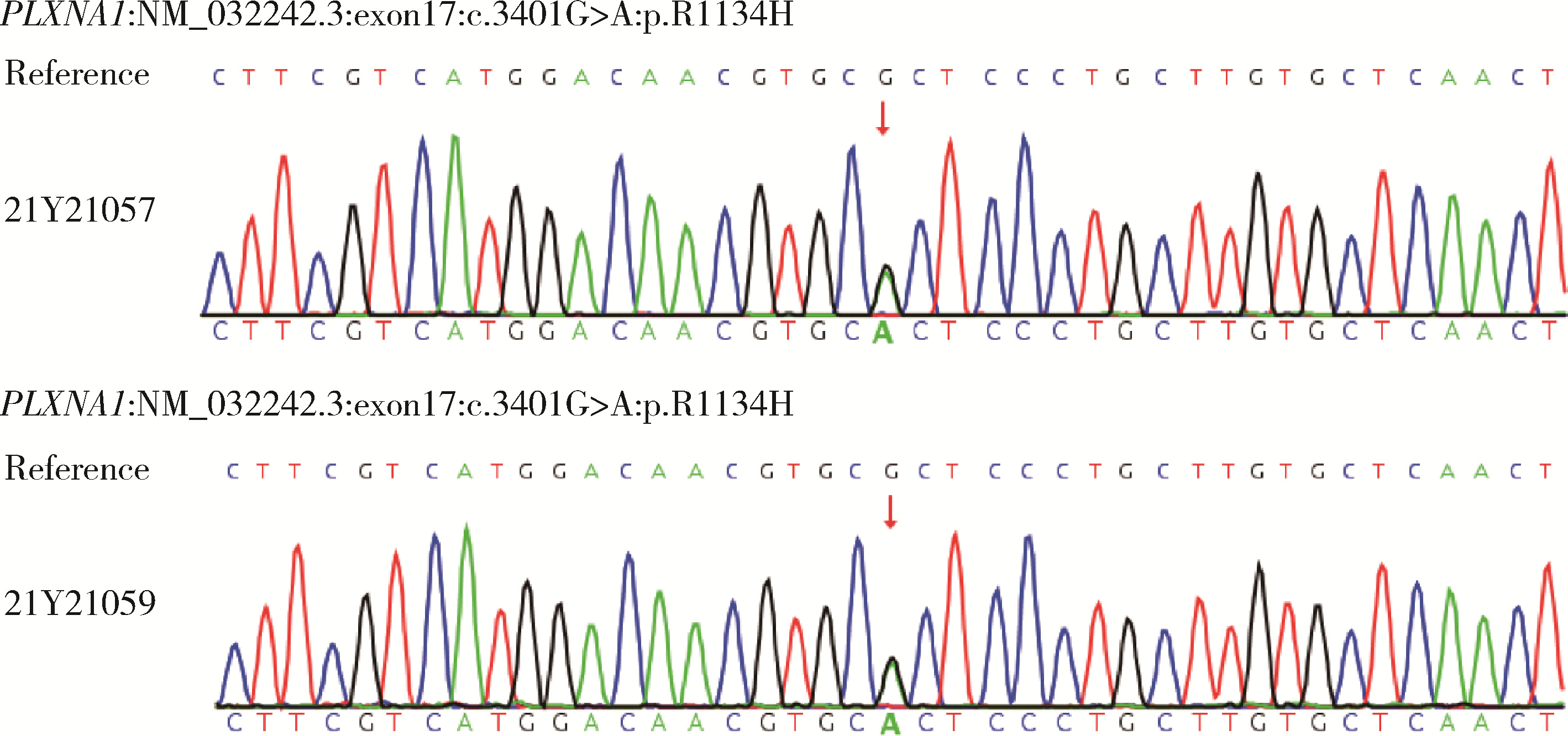
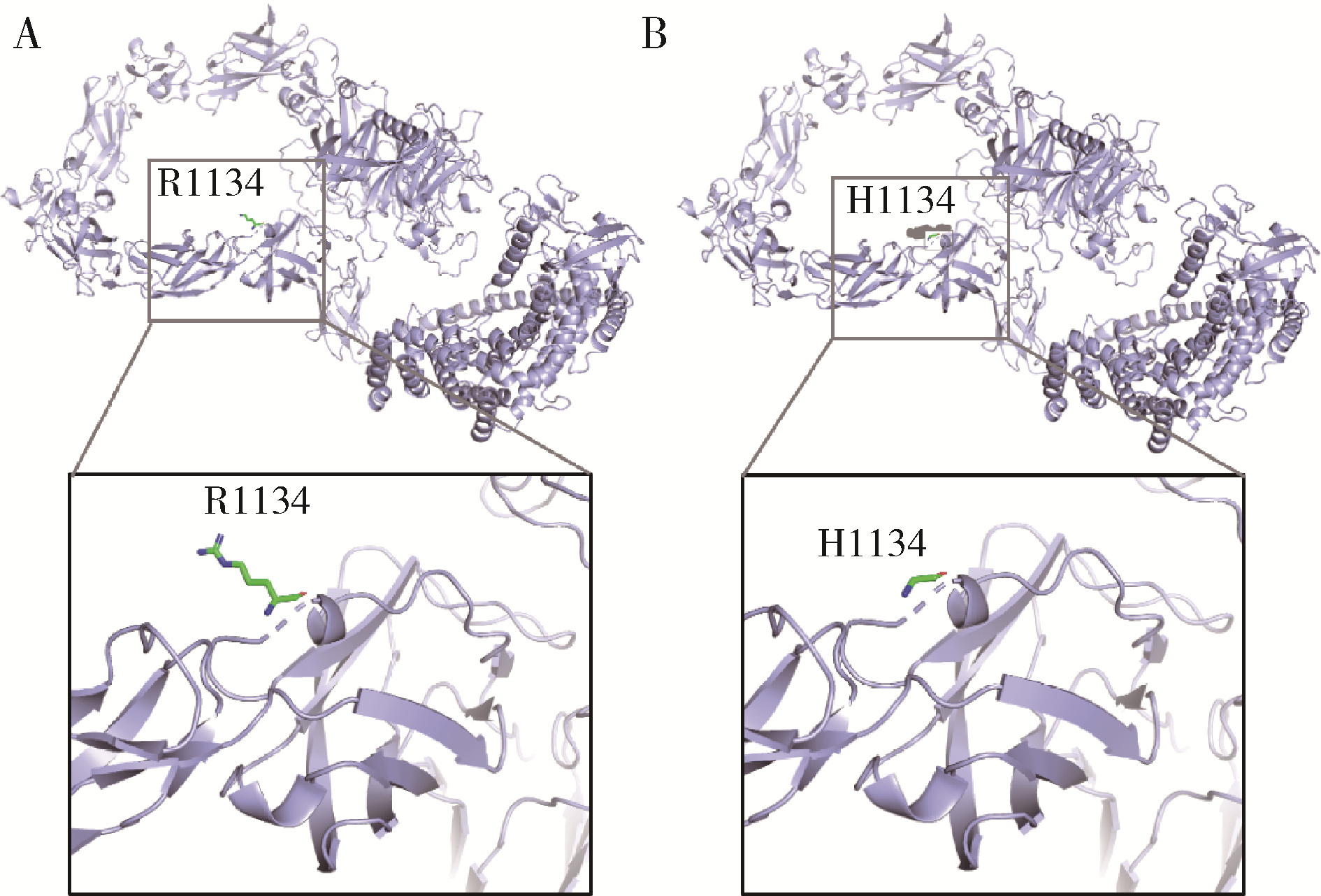
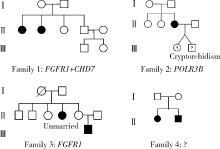
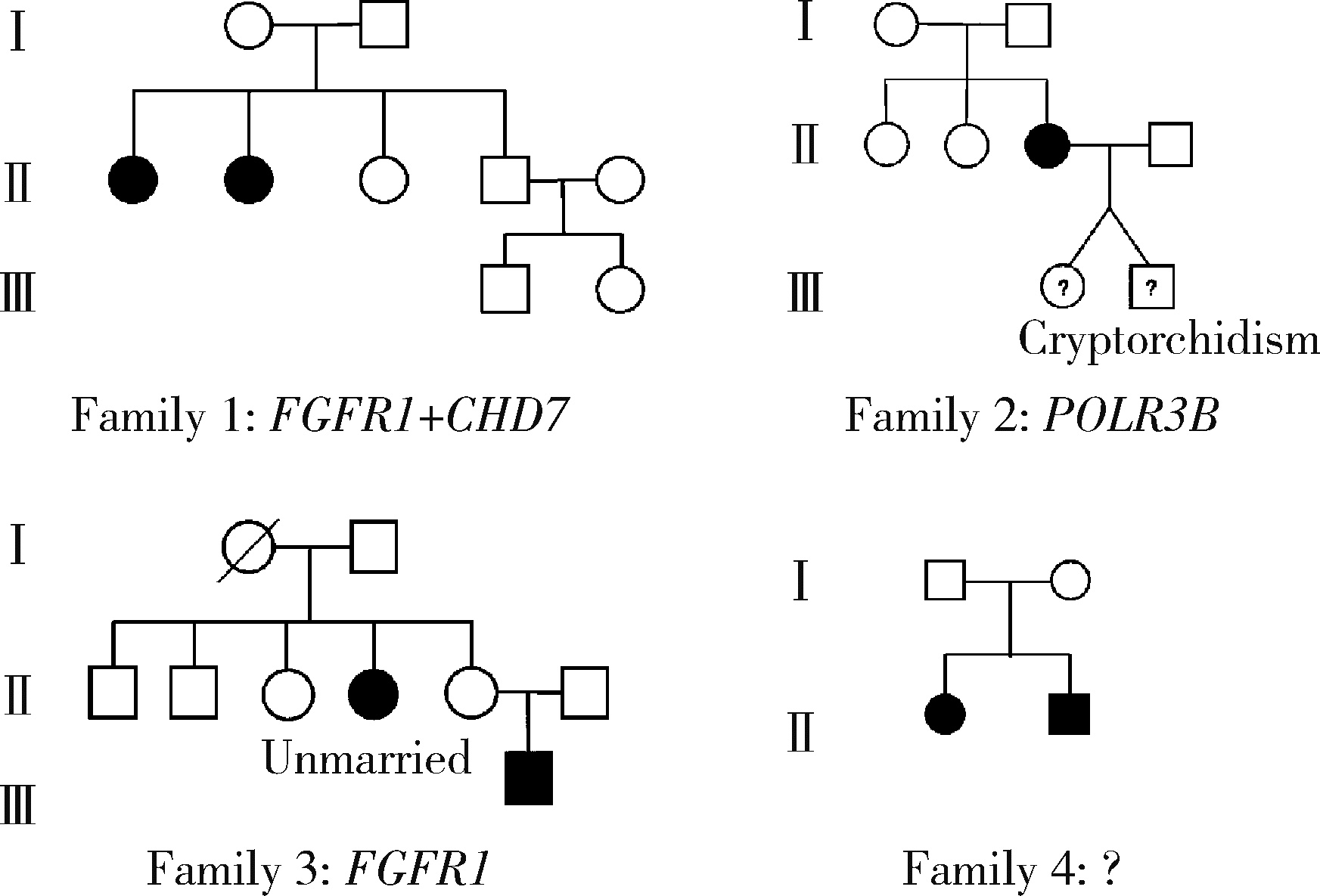
目的: 分析女性特发性低促性腺激素性腺功能减退症(idiopathic hypogonadotropic hypogonadism, IHH)患者的临床表型特点及遗传学检测资料, 探究其遗传学病因及遗传模式。方法: 招募21例女性IHH患者, 依据是否存在嗅觉异常分为非嗅觉异常IHH组及卡尔曼综合征(Kallmann syndrome, KS)组, 收集并分析其临床资料。通过全外显子组测序及Sanger测序分析遗传学病因, 包括已知疾病致病基因相关变异位点。针对PLXNA1错义变异使用Alphafold2进行突变体蛋白质结构预测。结果: 非嗅觉异常IHH组和KS组基线资料差异无统计学意义。21例患者中有17例患者及其家系成员进行外周血采集及测序, 其中4例检出明确致病性基因变异, 涉及FGFR1、PROKR2基因, 检出率为23.5%, 其余13例患者未获得明确遗传诊断。2例KS患者未检出已知致病性基因变异, 均杂合携带PLXNA1: c.3401G>A变异, 该错义变异导致1134氨基酸位点疏水性改变, 但不携带丛状蛋白通路其他基因变异位点。4例患者存在相关家族史, 检出携带IHH相关风险基因变异, 涉及FGFR1、CHD7、POLR3B基因。由于位点致病性评级仅为临床意义不明, 且不符合基因型-表型家系共分离, 所以部分患者未能明确遗传诊断。临床治疗方案主要包括应用激素替代药物维持第二性征及人工月经周期, 并使用促性腺激素进行促排卵治疗以获得生育力。结论: 女性IHH患者遗传学病因复杂且存在多基因致病遗传模式, 遗传家系患者及散发患者存在一定遗传风险; PLXNA1: c.3401G>A变异位点可能为KS风险变异位点。
中图分类号:
- R394.8

| 1 |
doi: 10.1186/1750-1172-6-41 |
| 2 |
doi: 10.3389/fendo.2022.965074 |
| 3 |
doi: 10.1111/cen.14822 |
| 4 |
doi: 10.1093/hmg/ddx080 |
| 5 |
doi: 10.1242/dev.176461 |
| 6 |
汪保安, 马晓莉, 邹效漫, 等. 女性特发性低促性腺激素性性腺功能减退症的临床评价及KAL1和FGFR1基因突变分析[J]. 军医进修学院学报, 2011, 32 (10): 1017- 1019.
|
| 7 |
曾旺, 李家大, 王新颖, 等. 中国先天性低促性腺激素性性腺功能减退症患者ANOS1的突变(英文)[J]. 中南大学学报(医学版), 2022, 47 (7): 847- 857.
|
| 8 |
张丽琼, 张红艳, 李明泸, 等. 低促性腺激素性性腺功能减退症患者KAL-1基因突变分析[J]. 云南医药, 2010, 31 (1): 27- 30.
|
| 9 |
解一丹, 郑瑞芝, 韩宾宾, 等. 22例低促性腺激素性性腺功能减退症患者CHD7基因变异分析[J]. 中华医学遗传学杂志, 2022, 39 (6): 571- 575.
|
| 10 |
doi: 10.1002/mgg3.1816 |
| 11 |
doi: 10.1111/cge.13482 |
| 12 |
|
| [1] | 谢尚,蔡志刚,单小峰. 全外显子测序及相关指标在口腔鳞状细胞癌精准治疗中的应用价值[J]. 北京大学学报(医学版), 2023, 55(4): 697-701. |
| [2] | 冯科,倪菁菁,夏彦清,曲晓伟,张慧娟,万锋,洪锴,张翠莲,郭海彬. 3例SUN5基因变异导致无头精子症的遗传学分析和助孕治疗结局[J]. 北京大学学报(医学版), 2021, 53(4): 803-807. |
| [3] | 柳小珍,李莹莹,杨丽萍. 全外显子组测序和目标序列靶向捕获测序在遗传性视网膜变性基因诊断中的差异[J]. 北京大学学报(医学版), 2020, 52(5): 836-844. |
| [4] | 邵为民, 白文俊, 陈益民, 刘磊, 王玉杰. 微量泵脉冲输注戈那瑞林治疗垂体柄中断综合征性腺功能减退病例分析及文献回顾[J]. 北京大学学报(医学版), 2014, 46(4): 642-645. |
| [5] | 陈亮, 徐阳 , 左文莉 , 杨慧霞, 廖秦平, 辛钟成, 郭应禄. Ras分子信号蛋白调控睾丸间质细胞Cox7a2蛋白表达及共定位影响研究[J]. 北京大学学报(医学版), 2012, 44(4): 507-510. |
| [6] | 李维仁*, 韩勃萱*, 刘涛, 李广永, 周峰, 巩艳青, 高喆珠, 崔万寿, 白光一, 辛钟成. 老年人睾丸间质细胞结构及StAR和P450scc蛋白表达的变化[J]. 北京大学学报(医学版), 2011, 43(4): 505-508. |
|
||
