北京大学学报(医学版) ›› 2021, Vol. 53 ›› Issue (5): 957-963. doi: 10.19723/j.issn.1671-167X.2021.05.025
远端型遗传性运动神经病8例的临床、病理及遗传学特点
刘梅歌1,方朴2,王严1,丛璐1,范洋溢1,袁远1,徐燕1,张俊1,洪道俊1,2,△( )
)
- 1.北京大学人民医院神经内科,北京 100044
2.南昌大学第一附属医院神经内科,南昌 330006
Clinical, pathological and genetic characteristics of 8 patients with distal hereditary motor neuropathy
LIU Mei-ge1,FANG Pu2,WANG Yan1,CONG Lu1,FAN Yang-yi1,YUAN Yuan1,XU Yan1,ZHANG Jun1,HONG Dao-jun1,2,△()
- 1. Department of Neurology, Peking University People’s Hospital, Beijing 100044, China
2. Department of Neurology, the First Affiliated Hospital of Nanchang University, Nanchang 330006, China
摘要:
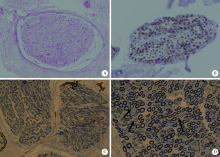
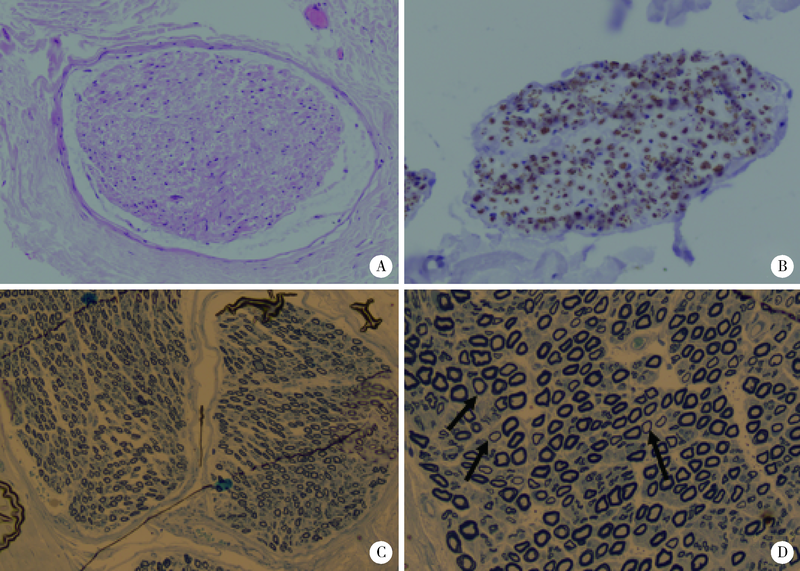
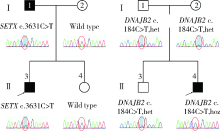
目的: 远端型遗传性运动神经病(distal hereditary motor neuropathy, dHMN)是一组选择性累及运动神经及其神经元的退行性病变,可引起肢体远端肌肉进行性萎缩无力。总结8例dHMN先证者的临床、电生理、病理及遗传学特点,丰富我国dHMN先证者的临床表型和基因型资料,提高临床工作者对dHMN的认识和诊治水平。方法: 选择2018年6月至2019年4月于北京大学人民医院神经内科就诊的8例dHMN先证者并进而追踪其家系,回顾性分析先证者的临床症状、神经电生理改变、病理特点及基因突变情况。运用基因靶向二代测序技术对所有先证者进行周围神经病相关基因检测,通过 Sanger测序验证突变位点,并对可获得的家系成员进行遗传共分离分析。结果: 先证者发病年龄11~64岁,中位数39.5岁,均为慢性起病,进行性发展,主要表现为远端肢体无力,并逐渐出现肌肉萎缩。神经电生理结果示选择性运动神经损害,运动神经复合肌肉动作电位波幅下降伴神经传导速度减慢,感觉神经不受累,针刺肌电图符合神经源性损害表现。2例先证者肌肉活检显示神经源性骨骼肌损害,1例先证者腓肠神经活检提示感觉神经受累轻微。基因测序显示8例先证者携带了8种不同的已知dHMN致病基因,3例有已报道的致病突变位点,基因诊断率为37.5%,其余5例为临床意义未明的新发点突变,其中2例突变在家系内共分离。结论: dHMN是一组临床和基因均具有显著异质性的遗传性周围神经病,二代测序技术广泛运用于dHMN先证者的致病基因搜寻,但仍有超过一半的先证者不能得到明确的基因诊断。
中图分类号:
- R596



| [1] |
Garg N, Park SB, Vucic S, et al. Differentiating lower motor neuron syndrome [J]. J Neurol Neurosurg Psychiatry, 2017, 88(6):474-483.
doi: 10.1136/jnnp-2016-313526 |
| [2] |
Frasquet M, Rojas-García R, Argente-Escrig H, et al. Distal hereditary motor neuropathies: mutation spectrum and genotype-phenotype correlation [J]. Eur J Neurol, 2021, 28(4):1334-1343.
doi: 10.1111/ene.14700 pmid: 33369814 |
| [3] |
Bansagi B, Griffin H, Whittaker RG, et al. Genetic heterogeneity of motor neuropathies [J]. Neurology, 2017, 88(13):1226-1234.
doi: 10.1212/WNL.0000000000003772 |
| [4] | Bacquet J, Stojkovic T, Boyer A, et al. Molecular diagnosis of inherited peripheral neuropathies by targeted next-generation sequencing: molecular spectrum delineation [J]. BMJ Open, 2018, 8(10):e21632. |
| [5] |
Echaniz-Laguna A, Geuens T, Petiot P, et al. Axonal neuropathies due to mutations in small heat shock proteins: clinical, genetic, and functional insights into novel mutations [J]. Hum Mutat, 2017, 38(5):556-568.
doi: 10.1002/humu.23189 pmid: 28144995 |
| [6] |
Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology [J]. Genet Med, 2015, 17(5):405-424.
doi: 10.1038/gim.2015.30 pmid: 25741868 |
| [7] |
Tanabe H, Higuchi Y, Yuan JH, et al. Clinical and genetic features of charcot-marie-tooth disease 2F and hereditary motor neuropathy 2B in Japan [J]. J Peripher Nerv Syst, 2018, 23(1):40-48.
doi: 10.1111/jns.2018.23.issue-1 |
| [8] |
Windpassinger C, Auer-Grumbach M, Irobi J, et al. Heterozygous missense mutations in BSCL2 are associated with distal hereditary motor neuropathy and Silver syndrome [J]. Nat Genet, 2004, 36(3):271-276.
doi: 10.1038/ng1313 |
| [9] |
Novarino G, Fenstermaker AG, Zaki MS, et al. Exome sequencing links corticospinal motor neuron disease to common neurodegenerative disorders [J]. Science, 2014, 343(6170):506-511.
doi: 10.1126/science.1247363 pmid: 24482476 |
| [10] | Xie Y, Lin Z, Pakhrin PS, et al. Genetic and clinical features in 24 Chinese distal hereditary motor neuropathy families [J/OL]. Front Neurol, 2020, 11:603003(2020-12-14)[2020-12-15]. https://pubmed-ncbi-nlm-nih-gov-443.webvpn.bjmu.edu.cn/33 3810781/ . |
| [11] | 张付峰, 卢晓琴, 严新翔, 等. 远端型遗传性运动神经病的临床特征分析 [J]. 第二军医大学学报, 2009, 30(1):57-60. |
| [12] |
De Jonghe P, Auer-Grumbach M, Irobi J, et al. Autosomal dominant juvenile amyotrophic lateral sclerosis and distal hereditary motor neuronopathy with pyramidal tract signs: synonyms for the same disorder [J]. Brain, 2002, 125(Pt 6):1320-1325.
pmid: 12023320 |
| [13] |
Motley WW, Griffin LB, Mademan I, et al. A novel AARS mutation in a family with dominant myeloneuropathy [J]. Neurology, 2015, 84(20):2040-2047.
doi: 10.1212/WNL.0000000000001583 |
| [14] |
Luigetti M, Fabrizi GM, Madia F, et al. Seipin S90L mutation in an Italian family with CMT2/dHMN and pyramidal signs [J]. Muscle Nerve, 2010, 42(3):448-451.
doi: 10.1002/mus.21734 pmid: 20806400 |
| [15] |
Beecroft SJ, McLean CA, Delatycki MB, et al. Expanding the phenotypic spectrum associated with mutations of DYNC1H1 [J]. Neuromuscul Disord, 2017, 27(7):607-615.
doi: 10.1016/j.nmd.2017.04.011 |
| [16] |
Dierick I, Baets J, Irobi J, et al. Relative contribution of mutations in genes for autosomal dominant distal hereditary motor neuropathies: a genotype-phenotype correlation study [J]. Brain, 2008, 131(Pt 5):1217-1227.
doi: 10.1093/brain/awn029 pmid: 18325928 |
| [17] |
Rossor AM, Evans MR, Reilly MM. A practical approach to the genetic neuropathies [J]. Pract Neurol, 2015, 15(3):187-198.
doi: 10.1136/practneurol-2015-001095 |
| [18] |
Liu X, Duan X, Zhang Y, Sun A, et al. Molecular analysis and clinical diversity of distal hereditary motor neuropathy [J]. Eur J Neurol, 2020, 27:1319-1326.
doi: 10.1111/ene.14260 pmid: 32298515 |
| [19] |
Beijer D, Baets J. The expanding genetic landscape of hereditary motor neuropathies [J]. Brain, 2020, 143(Pt 12):3540-3563.
doi: 10.1093/brain/awaa311 |
| [1] | 吴婧娴, 郑柳燕, 于欢, 王淮蓉, 解舒婷, 陈雅霖, 李腾, 王梦莹, 秦雪英, 吴涛, 陈大方, 武轶群, 胡永华. CTNNA基因家族遗传培育效应与缺血性脑卒中的关联分析[J]. 北京大学学报(医学版), 2026, 58(3): 528-535. |
| [2] | 白红娟, 陈洋, 王琨, 徐学兵. 家庭创伤、校园霸凌与青少年自杀行为的关系:DRD2基因多态性的调节作用[J]. 北京大学学报(医学版), 2026, 58(3): 606-615. |
| [3] | 高鹏, 刘燕玲, 李俊峰, 姚立琼. B/C基因型的乙型肝炎病毒在不同丙氨酸氨基转移酶状态下对Th17、Treg细胞及其比率的影响[J]. 北京大学学报(医学版), 2026, 58(3): 674-677. |
| [4] | 付浩, 申潞艳, 黄冰洋, 马少华. 免疫治疗背景下食管鳞状细胞癌围手术期治疗的临床思考[J]. 北京大学学报(医学版), 2026, 58(2): 266-271. |
| [5] | 殷昊明, 王子杰, 舒帆, 张展奕, 梁会, 张树栋. 肾透明细胞癌FABP6基因长转录本的表达及意义[J]. 北京大学学报(医学版), 2026, 58(2): 393-398. |
| [6] | 耿芸玲, 刘超, 杨萍, 郑佳佳, 沈宁, 杜毅鹏. 医院获得性肺炎患者肺炎克雷伯菌多部位感染的临床特征及毒力基因分布[J]. 北京大学学报(医学版), 2026, 58(1): 201-207. |
| [7] | 刘艳华, 陆敏, 赵旭阳, 张宽根, 武睿, 梅放, 戴志豪, 由江峰, 裴斐. 肿瘤转移抑制基因LASS2去磷酸化对液泡型ATP酶活性及前列腺癌侵袭性的影响[J]. 北京大学学报(医学版), 2025, 57(6): 1113-1123. |
| [8] | 董琪, 何菁, 贾园, 姚海红, 张霞. 模拟复发性多软骨炎的VEXAS综合征1例[J]. 北京大学学报(医学版), 2025, 57(6): 1180-1183. |
| [9] | 王晓林, 李璐瑶, 张雯, 王鸿雁. 3例子宫体中肾样腺癌的临床病理学分析[J]. 北京大学学报(医学版), 2025, 57(6): 1208-1212. |
| [10] | 王小梦, 曾晓君, 李娟. 黎族与汉族系统性红斑狼疮患者的主要临床特征[J]. 北京大学学报(医学版), 2025, 57(6): 1213-1218. |
| [11] | 张展奕, 陆敏, 孙悦皓, 董靖晗, 侯小飞, 肖春雷, 王国良, 田晓军, 马潞林, 张洪宪, 张树栋. TFE3重排肾细胞癌合并静脉癌栓患者的临床病理特征及生存分析[J]. 北京大学学报(医学版), 2025, 57(4): 650-661. |
| [12] | 周泽臻, 葛力源, 张帆, 邓绍晖, 颜野, 张洪宪, 王国良, 刘磊, 黄毅, 张树栋. 病理T3a期肾细胞癌肾部分切除与根治性肾切除的回顾性匹配研究[J]. 北京大学学报(医学版), 2025, 57(4): 704-710. |
| [13] | 李梦迪, 雷蕾, 刘中宁, 李健, 姜婷. siRNA沉默NLK基因促进神经化组织工程骨再生[J]. 北京大学学报(医学版), 2025, 57(2): 227-236. |
| [14] | 张真伟, 徐欣然, 高学军, 董艳梅, 田华. RELT基因移码突变导致遗传性釉质发育不全[J]. 北京大学学报(医学版), 2025, 57(1): 13-18. |
| [15] | 马民英, 晁晓芹, 赵扬, 赵国廷. LncRNA SNHG20靶向调控miR-520c-3p/RAB22A通路对人口腔鳞状细胞癌细胞上皮间质转化及微管形成的影响[J]. 北京大学学报(医学版), 2025, 57(1): 26-32. |
|
||
