北京大学学报(医学版) ›› 2023, Vol. 55 ›› Issue (1): 181-185. doi: 10.19723/j.issn.1671-167X.2023.01.028
1例Angelman综合征合并眼皮肤白化病2型患者的临床和遗传学分析及文献回顾
周秋君,龚潘,焦莶如,杨志仙*( )
)
- 北京大学第一医院儿科,北京 100034
Clinical and molecular genetic analysis of Angelman syndrome with oculocutaneous albinism type 2: A case report and literature review
Qiu-jun ZHOU,Pan GONG,Xian-ru JIAO,Zhi-xian YANG*()
- Department of Pediatrics, Peking University First Hospital, Beijing 100034, China
摘要:
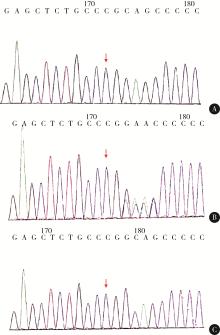
总结1例Angelman综合征(Angelman syndrome, AS)合并眼皮肤白化病2型(oculocutaneous albinism type 2, OCA2)患儿的临床诊疗过程及遗传学检测结果和特点,并以“Angelman综合征”“眼皮肤白化病2型”“Angelman syndrome”“P gene”“Oculocutaneous albinism type 2”为关键词分别在CNKI、万方数据库及PubMed数据库(自建库至2019年12月)中检索,对国内外报道的AS合并OCA2病例进行汇总分析。本例患儿女,1岁,出生后即发现全身白,毛发色黄,眼球震颤,2个月会竖头,7个月会翻身,测头围42 cm,不能独坐,不会说话。家系全外显子组基因测序显示,患儿携带P基因c.168del(p.Gln58ArgfsTer44)纯合突变,经验证其父亲为杂合型,母亲为野生型。拷贝数变异检测提示,患儿母源染色体15q11.2-13.1区域缺失(P基因位于此区域内)。截至2019年12月,3个数据库中共检索到4篇相关文献,共报道了4例AS合并OCA2患儿,与本例一起进行汇总分析。AS合并OCA2患儿出生后均表现出皮肤白、毛发金黄、虹膜颜色浅,出生后6月龄左右发现全面发育迟缓,有2例随访至儿童期,语言始终无发育。4例患儿病程中出现癫痫发作,2例有共济失调,5例均有获得性小头畸形,2例白化病家族史阳性,3例完成脑电图监测结果均异常。遗传学检测结果显示,5例患儿均为母源性染色体15q11-13区域缺失,4例有父源15号染色体P基因突变,1例未进行P基因检测而根据临床诊断OCA2。AS合并OCA2病例相对少见,根据出生后明显的临床表现较容易获得OCA2临床诊断,当合并神经发育迟缓等临床表现时,提示早期临床难以诊断的AS可能,遗传学检测两者的交叉遗传现象可最终确诊此种复合病。
中图分类号:
- R596



| 1 | Williams CA , Beaudet AL , Clayton-Smith J , et al. Angelman syndrome 2005: Updated consensus for diagnostic criteria[J]. Am J Med Genet A, 2006, 140 (5): 413- 418. |
| 2 | Spritz RA . Molecular genetics of oculocutaneous albinism[J]. Semin dermatol, 1993, 12 (3): 167- 172. |
| 3 |
Saitoh S , Oiso N , Wada T , et al. Oculocutaneous albinism type 2 with a P gene missense mutation in a patient with Angelman syndrome[J]. J Med Genet, 2000, 37 (5): 392- 394.
doi: 10.1136/jmg.37.5.392 |
| 4 |
Saadeh R , Lisi EC , Batista DAS , et al. Albinism and developmental delay: The need to test for 15q11-q13 deletion[J]. Pediatr Neurol, 2007, 37 (4): 299- 302.
doi: 10.1016/j.pediatrneurol.2007.06.024 |
| 5 |
Fridman C , Hosomi N , Varela MC , et al. Angelman syndrome associated with oculocutaneous albinism due to an intragenic deletion of the P gene[J]. Am J Med Genet A, 2003, 119A (2): 180- 183.
doi: 10.1002/ajmg.a.20105 |
| 6 |
李洪义, 郑辉. Angelman综合征并眼皮肤白化病一例[J]. 中华儿科杂志, 2005, 43 (8): 635- 636.
doi: 10.3760/j.issn:0578-1310.2005.08.028 |
| 7 |
Holm VA , Cassidy SB , Butler MG , et al. Prader-Willi syndrome: Consensus diagnostic criteria[J]. Pediatrics, 1993, 91 (2): 398- 402.
doi: 10.1542/peds.91.2.398 |
| 8 |
Oetting WS , King RA . Molecular basis of albinism: Mutations and polymorphisms of pigmentation genes associated with albinism[J]. Hum Mutat, 1999, 13 (2): 99- 115.
doi: 10.1002/(SICI)1098-1004(1999)13:2<99::AID-HUMU2>3.0.CO;2-C |
| 9 |
Biswas S , Lloyd IC . Oculocutaneous albinism[J]. Arch Dis Child, 1999, 80 (6): 565- 569.
doi: 10.1136/adc.80.6.565 |
| 10 | 李洪义, 吴维青, 郑辉. 眼皮肤白化病常见亚型的基因与基因突变[J]. 中国优生与遗传杂志, 2004, 12 (1): 118- 121. |
| 11 |
段红蕾, 郑辉, 李洪义. 眼皮肤白化病Ⅱ型相关的P基因突变与DNA多态性[J]. 遗传, 2005, 27 (6): 984- 985.
doi: 10.3321/j.issn:0253-9772.2005.06.023 |
| 12 |
Gronskov K , Ek J , Brondum-Nielsen K . Oculocutaneous albinism[J]. Orphanet J Rare Dis, 2007, 2 (1): 43- 45.
doi: 10.1186/1750-1172-2-43 |
| 13 | 李璞. 医学遗传学[M]. 北京: 北京医科大学中国协和医科大学联合出版社, 1999: 60. |
| 14 |
Carden SM , Boissy RE , Schoettker PJ , et al. Albinism: Modern molecular diagnosis[J]. Br J Ophthalmol, 1998, 82 (2): 189- 195.
doi: 10.1136/bjo.82.2.189 |
| 15 |
Clayton-Smith J , Webb T , Robb SA , et al. Further evidence for dominant inheritance at the chromosome 15q11-13 locus in familial Angelman syndrome[J]. Am J Med Genet, 1992, 44 (2): 256- 260.
doi: 10.1002/ajmg.1320440236 |
| 16 |
Uemura N , Matsumoto A , Nakamura M , et al. Evolution of seizures and electroencephalographical findings in 23 cases of deletion type Angelman syndrome[J]. Brain Dev, 2005, 27 (5): 383- 388.
doi: 10.1016/j.braindev.2004.01.009 |
| 17 | 杨志仙, 刘晓燕, 秦炯, 等. Angelman综合征临床及脑电图特征[J]. 中国实用儿科杂志, 2011, 26 (7): 519- 523. |
| 18 |
Thibert RL , Conant KD , Braun EK , et al. Epilepsy in Angelman syndrome: A questionnaire-based assessment of the natural history and current treatment options[J]. Epilepsia, 2009, 50 (11): 2369- 2376.
doi: 10.1111/j.1528-1167.2009.02108.x |
| [1] | 舒帆, 葛力源, 邓汉彰, 殷昊明, 欧俊永, 邓绍晖, 郝一昌, 陆敏, 张展奕, 段佩辰, 张树栋. 预后不良的肾细胞癌伴淋巴结转移的分子特征[J]. 北京大学学报(医学版), 2026, 58(3): 631-640. |
| [2] | 罗必显, 刘洪铭, 谢伟勋, 龚渭华. 产甲胎蛋白胃癌的新临床特征和前沿科学问题[J]. 北京大学学报(医学版), 2026, 58(2): 257-265. |
| [3] | 董琪, 何菁, 贾园, 姚海红, 张霞. 模拟复发性多软骨炎的VEXAS综合征1例[J]. 北京大学学报(医学版), 2025, 57(6): 1180-1183. |
| [4] | 张真伟, 徐欣然, 高学军, 董艳梅, 田华. RELT基因移码突变导致遗传性釉质发育不全[J]. 北京大学学报(医学版), 2025, 57(1): 13-18. |
| [5] | 金银姬, 刘蕊. 以肠系膜静脉血栓为突出表现的遗传性蛋白S缺乏症1例[J]. 北京大学学报(医学版), 2024, 56(6): 1106-1109. |
| [6] | 时云飞,王豪杰,刘卫平,米岚,龙孟平,刘雁飞,赖玉梅,周立新,刁新婷,李向红. 血管免疫母细胞性T细胞淋巴瘤临床与分子病理学特征分析[J]. 北京大学学报(医学版), 2023, 55(3): 521-529. |
| [7] | 熊焰,张波,聂立功,吴世凯,赵虎,李东,邸吉廷. 胸部SMARCA4缺失性未分化肿瘤的病理诊断与联合免疫检测点抑制剂治疗[J]. 北京大学学报(医学版), 2023, 55(2): 351-356. |
| [8] | 程晓静,蒋栋,张连海,王江华,李雅真,翟佳慧,闫宝琪,张露露,谢兴旺,李子禹,季加孚. KRAS G12V特异性T细胞受体治疗恶性肿瘤的临床前研究[J]. 北京大学学报(医学版), 2022, 54(5): 884-895. |
| [9] | 秦彩朋,宋宇轩,丁梦婷,王飞,林佳兴,杨文博,杜依青,李清,刘士军,徐涛. 肾癌免疫治疗疗效评估突变预测模型的建立[J]. 北京大学学报(医学版), 2022, 54(4): 663-668. |
| [10] | 陈曦,王斯悦,薛恩慈,王雪珩,彭和香,范梦,王梦莹,武轶群,秦雪英,李劲,吴涛,朱洪平,李静,周治波,陈大方,胡永华. 基于核心家系全外显子组测序数据探索新生突变与非综合征型唇腭裂的关联[J]. 北京大学学报(医学版), 2022, 54(3): 387-393. |
| [11] | 曹泽,王乐童,刘振明. 严重急性呼吸综合征冠状病毒2的Spike蛋白点突变后与受体蛋白质及潜在抗病毒药物结合能力的同源建模分析[J]. 北京大学学报(医学版), 2021, 53(1): 150-158. |
| [12] | 吴君怡,余淼,孙仕晨,樊壮壮,郑静蕾,张刘陶,冯海兰,刘洋,韩冬. 少汗性外胚层发育不良患者EDA基因突变检测及表型分析[J]. 北京大学学报(医学版), 2021, 53(1): 24-33. |
| [13] | 鲍轶,莫娟芬. 同时性多原发肺腺癌组织编码转录因子ERG基因相同位点突变1例报告[J]. 北京大学学报(医学版), 2020, 52(5): 971-974. |
| [14] | 张宽根,周雨禾,邵雅昆,梅放,由江峰,刘北英,裴斐. 肿瘤转移抑制基因LASS2/TMSG1 S248A突变体通过增加ATP6V0C表达促进前列腺癌的侵袭[J]. 北京大学学报(医学版), 2019, 51(2): 210-220. |
| [15] | 王皓,刘洋,刘浩辰,韩冬,冯海兰. 先天性缺牙患者中BMP2基因突变检测及功能分析[J]. 北京大学学报(医学版), 2019, 51(1): 9-15. |
|
||
